Contact Chercheur
Hugues Roest Crollius
Chercheur CNRS
01.44.32.23.70
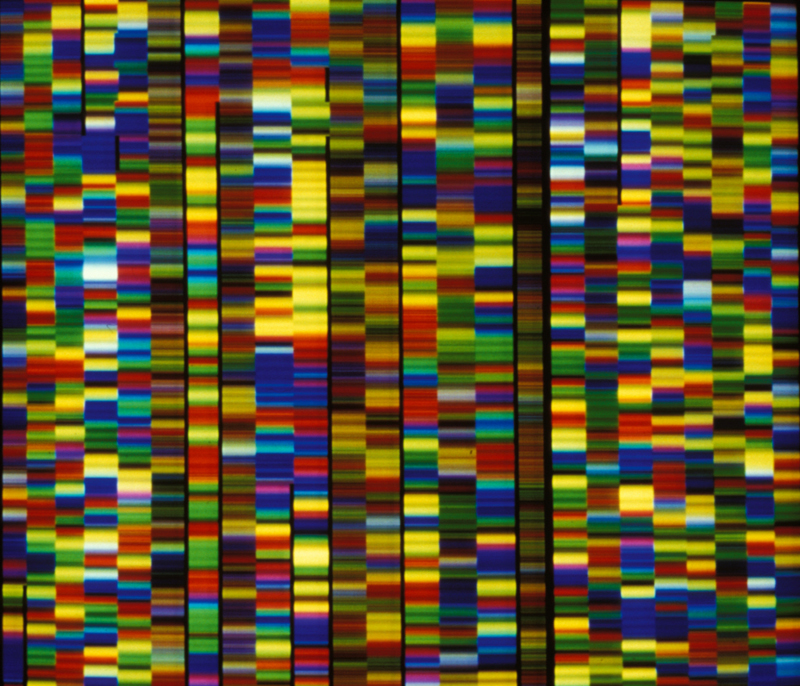
© Inserm, B. Jordan L’ADN est une longue suite de 3 milliards de « lettres », qui sont quatre petites molécules : l’adénine (A), la thymine (T), la guanine (G) et la cytosine (C). Nos gènes sont l’équivalent de mots, écrits, dans toutes les espèces vivantes, à partir de ces quatre lettres.
Les chercheurs de l’équipe Dynamique et organisation des génomes, à l’Institut de biologie de l’Ecole normale supérieure (CNRS/ENS/Inserm) ont étudié le génome de l’homme et de trois autres primates (chimpanzé, orang-outan et le macaque) grâce à des outils bioinformatiques. Leur travail a consisté à comparer ces génomes entiers pour identifier les gènes sélectionnés au cours des derniers 200 000 ans dans chaque espèce. Résultat : quelques centaines de gènes ont été récemment sélectionnés chez chacune de ces espèces. Parmi eux, environ 100 gènes détectés chez l’homme sont partagés par deux des trois autres espèces, soit deux fois plus qu’attendu du simple fait du hasard (1).
Cette étude permet aussi de mieux cerner le groupe de gènes spécifiquement mis en jeu au cours de l’évolution chez l’homme (pendant les derniers 200 000 ans), puisque l’on sait maintenant lesquels n’ont été sélectionnés dans aucune autre lignée de primates. C’est le cas déjà bien connu, et que cette étude confirme, du gène de la lactase, qui permet de métaboliser le lactose du lait à l’âge adulte (avantage certain avec l’apparition de l’agriculture et de l’élevage). Les chercheurs ont également identifié un groupe de gènes impliqués dans certaines fonctions neurologiques et dans le développement des muscles et du squelette.
Le niveau de variabilité comme indicateur de la sélection
Jusqu’à présent, l’identification des gènes sélectionnés nécessitait de travailler sur les génomes de plusieurs dizaines d’individus suivant des méthodes statistiques. Elle n’avait été réalisée que chez l’homme. Les chercheurs ont mis au point une méthode ne nécessitant de disposer du génome que d’un seul individu. Elle est fondée sur la recherche des régions du génome très pauvres en polymorphisme allélique. Explications : chaque gène est présent dans le génome en deux exemplaires, que l’on appelle allèles (un sur chaque chromosome) et qui ne sont pas parfaitement identiques : il existe un certain polymorphisme. Lorsqu’une mutation avantageuse se produit et qu’elle se répand dans toute la population, le génome de chaque individu devient identique dans la région entourant le gène concerné. Le polymorphisme est alors très faible (3): une mutation avantageuse a été sélectionnée au détriment de la variabilité locale du génome.
Reste à déterminer, à l’aide d’un plus grand nombre de génomes de primates, l’étendue de ce phénomène en termes de gènes et de fonctions biologiques. En incluant d’autres espèces de vertébrés dans l’étude, il sera également possible de déterminer si nous partageons des événements adaptatifs avec les rongeurs, les oiseaux ou les poissons, comme semblent déjà le suggérer quelques observations isolées.
(1) Ce résultat est même probablement sous-évalué, du fait du « bruit de fond » que génère la méthode employée.
(2) Par exemple, la résistance à certains virus chez les primates.
(3) Les chercheurs travaillent sur le rapport entre polymorphisme (nombre de bases différentes entre les 2 allèles) et divergence avec une espèce voisine (nombre de bases différentes avec cette espèce), ce afin de s’assurer que le faible polymorphisme n’est pas dû à une autre cause qu’une mutation avantageuse.
Hugues Roest Crollius
Chercheur CNRS
01.44.32.23.70
Enard, D., Depaulis, F., Roest Crollius, H. (2010)
"Human and non-human primate genomes share hotspots of positive selection"
Plos Genetics 6(2): e1000840. doi:10.1371/journal.pgen.1000840