Une équipe coordonnée par Antoine Triller, directeur de recherche Inserm, directeur de l’Institut de Biologie de l’Ecole Normale Supérieure, et Ronald Melki, directeur de recherche CNRS (Institut des Neurosciences de Paris-Saclay), vient d’identifier la cible d’une protéine l’alpha-synucléine, qui est pathogène dans la maladie de Parkinson. Cette cible est une pompe sodium/potassium ATP-dépendante. Elle peut potentiellement être utilisée pour la mise au point de traitements symptomatiques de la maladie de Parkinson. Le détail de ces travaux est publié dans The EMBO Journal daté du 31 août 2015.
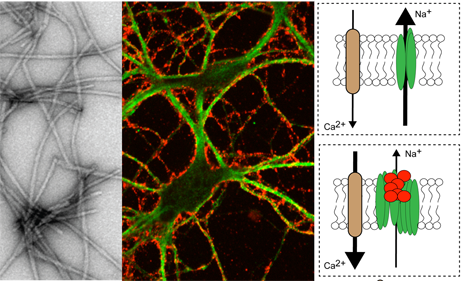
L’alpha-synucléine forme des fibrilles (en gris) qui se collent (en rouge) sur la membrane des neurones (en vert). Sur la partie droite de la figure : les fibrilles (en rouge), en s’agrégeant, perturbent le fonctionnement de la pompe (en vert) qui maintient le gradient de sodium (Na+). Cela dépolarise le neurone et augmente l’entrée de calcium (Ca2+) qui est toxique pour le neurone.© Inserm/Antoine Triller
L’alpha-synucléine fait partie (avec les protéines tau et bêta amyloïde pour la maladie d’Alzheimer, ou la protéine prion pour la maladie de Kreutzfeld-Jacob,) des protéines pathogènes qui se propagent de cellules en cellules et qui sont associées aux changements physiopathologiques observés dans les maladies neurodégénératives.
Antoine Triller et ses collègues ont montré que cette protéine s’agrège sur la membrane des neurones, et interagit avec une protéine de surface du neurone, la sous unité α3 de la pompe sodium (Na+)/potassium (K+) ATPase. Cette pompe contrôle les flux d’ions sodium et potassium dans les neurones, et par voie de conséquence, l’activité électrique de ces neurones.
Chez l’homme, des mutations de cette pompe sont responsables de symptômes moteurs de la maladie de Parkinson à début précoce et de l’hémiplégie alternante de l’enfant (HAE). Les chercheurs viennent de démontrer que l’apha-synucléine, qui diffuse entre les cellules, interagit avec la pompe Na+/K+ ATPase dans la membrane. La pompe, lorsqu’elle est liée à l’alpha-synucléine est moins à même d’effectuer son activité de pompage. L’excitabilité neuronale est perturbée. Peu à peu, les signaux entre neurones ne sont pas transmis normalement et les symptômes de la maladie de Parkinson ou de l’HAE apparaissent.
Cette découverte a été rendue possible grâce à la combinaison de techniques de biologie moléculaire et de microscopie super-résolutive permettant le suivi des molécules individuelles. Cette dernière approche a été couronnée en 2014 par le prix Nobel de chimie attribué à Eric Betzig, Stephan W. Hell et William E. Moerner.