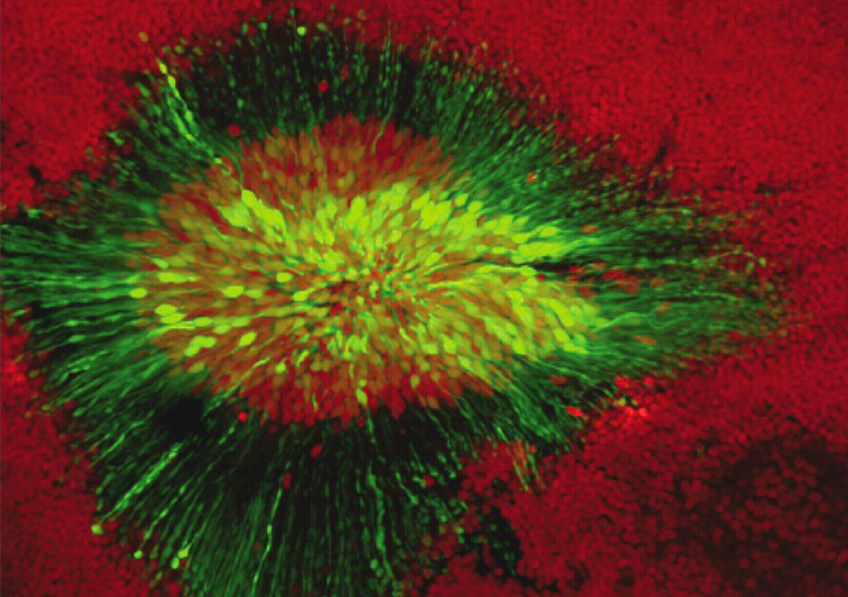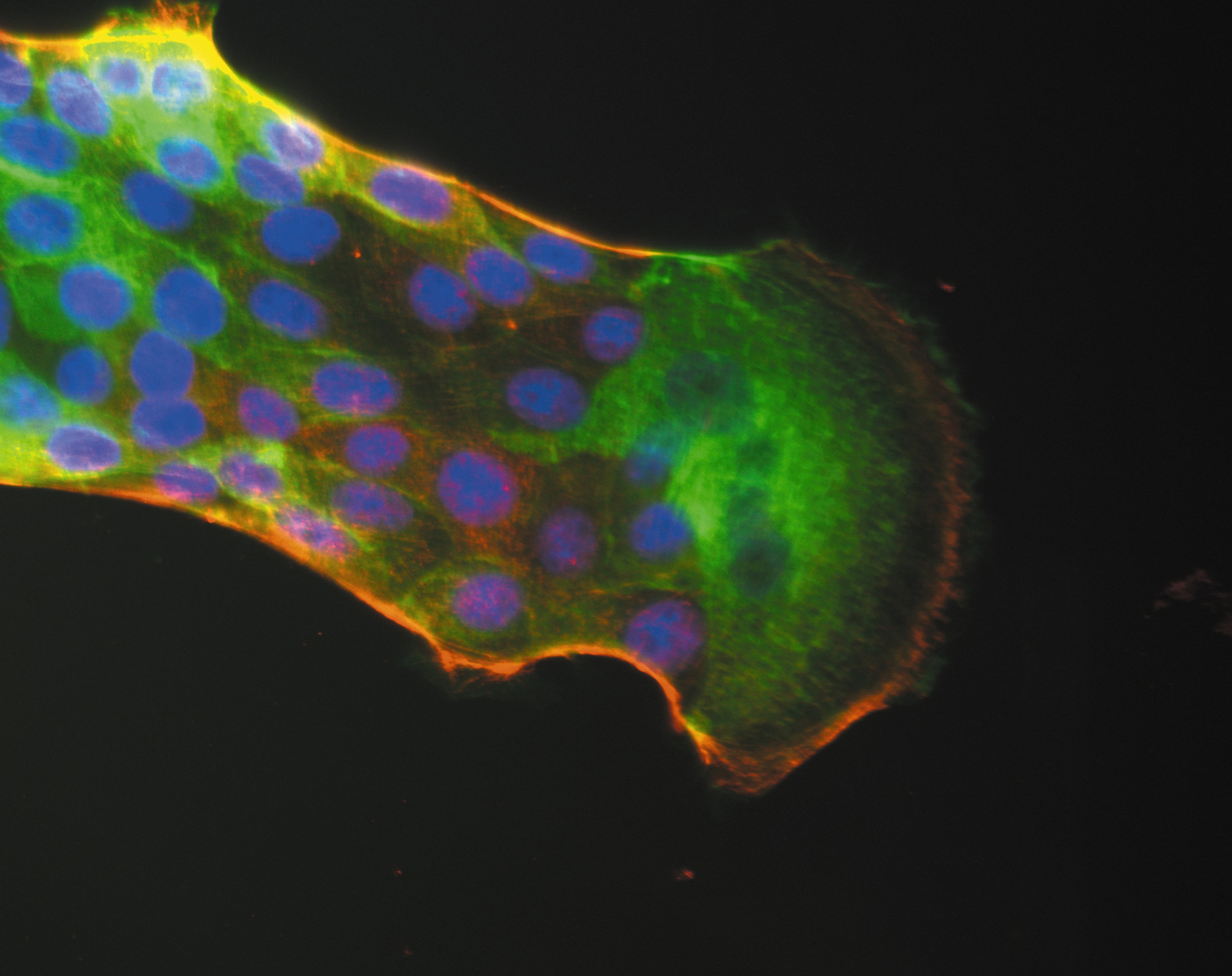
Doigt de migration cellulaire précédé par une cellule leader. En bleu, les noyaux des cellules, en vert, l’actine, en rouge, la myosine. Le câble pluricellulaire d’acto-myosine est bien visible sur les bords du doigt. ©Inserm/Cochet-Escartin, Olivier, 2014
L’asymétrie joue un rôle majeur en biologie, à toutes les échelles : enroulement en spirale de l’ADN, cœur positionné à gauche, préférence pour la main gauche ou la droite… Une équipe de l’Institut de biologie Valrose (CNRS/Inserm/Université Côte d’Azur), en collaboration avec des collègues de l’université de Pennsylvanie, a montré qu’une unique protéine induit le mouvement en spirale d’une autre molécule puis, par effet domino, la torsion des cellules, des organes et du corps entier, jusqu’à déclencher un comportement latéralisé. Ces travaux sont publiés dans la revue Science le 23 novembre 2018.
Notre monde est fondamentalement asymétrique : enroulement de la double hélice d’ADN, division asymétrique des cellules souches, localisation du cœur humain à gauche… Mais comment émergent ces asymétries et sont-elles liées les unes aux autres ?
À l’Institut de biologie Valrose l’équipe du chercheur CNRS Stéphane Noselli comprenant aussi des chercheurs de l’Inserm et de l’Université Cote d’Azur étudie depuis plusieurs années l’asymétrie droite-gauche afin de résoudre ces énigmes. Ces biologistes avaient identifié le premier gène contrôlant cette asymétrie chez la mouche du vinaigre (drosophile), l’un des organismes modèles préférés des biologistes. Plus récemment, l’équipe a montré que ce gène joue le même rôle chez les vertébrés : la protéine qu’il produit, la myosine 1D[1], contrôle l’enroulement ou la rotation des organes dans le même sens.
Dans cette nouvelle étude, les chercheurs ont induit la production de myosine 1D dans des organes normalement symétriques de la drosophile, comme les trachées respiratoires. De façon spectaculaire, cela a suffi à induire une asymétrie à tous les niveaux : cellules déformées, trachées s’enroulant sur elles-mêmes, organisme entier torsadé, et comportement de nage hélicoïdale des larves de mouches. Chose remarquable, ces nouvelles asymétries se développent toujours dans le même sens.
Afin d’identifier l’origine de ces effets en cascade, des biochimistes de l’université de Pennsylvanie ont apporté leur concours : ils ont mis en présence, sur une lame de verre, la myosine 1D et un composant du « squelette » des cellules, l’actine. Ils ont alors pu constater que l’interaction des deux protéines entraine un mouvement en spirale de l’actine.
Outre son rôle dans l’asymétrie droite-gauche chez la drosophile et les vertébrés, la myosine 1D apparaît donc comme une protéine unique capable à elle seule d’induire l’asymétrie à toutes les échelles, d’abord au niveau moléculaire, puis, par effet domino, cellulaire, tissulaire et comportemental.
Ces résultats suggèrent un mécanisme possible d’apparition soudaine de nouveaux caractères morphologiques au cours de l’évolution, comme par exemple la torsion du corps des escargots. La myosine 1D aurait toutes les caractéristiques requises pour l’émergence de cette innovation, puisque son expression suffit à elle seule à induire la torsion à toutes les échelles.[1] Les myosines sont une classe de protéines qui interagissent avec l’actine (constituant du squelette des cellules ou cytosquelette). La plus connue d’entre elles, la myosine musculaire, est responsable de la contraction musculaire.