Connue pour être responsable de la maladie de Huntington, une maladie neurodégénérative, la protéine huntingtine mutée est également impliquée dans la progression et l’agressivité des tumeurs mammaires. C’est le résultat des études menées par l’équipe de Sandrine Humbert[1], directrice de recherche Inserm à l’Institut Curie, et publiées le 9 janvier 2013 dans le journal EMBO Molecular Medicine. Au niveau cellulaire, la protéine huntingtine mutée empêche le bon fonctionnement du récepteur de type HER2[2] dont la surexpression conduit à une multiplication des cellules tumorales et à une survenue plus fréquente des métastases.
L’équipe de Sandrine Humbert montre pour la première fois que la protéine huntingtine est exprimée dans le tissu mammaire sain ainsi que dans les tumeurs mammaires. « De plus nous révélons que l’expression de la huntingtine mutante dans les tumeurs mammaires rend la tumeur plus agressive, en particulier parce que celle-ci développe davantage de métastases » explique Sandrine Humbert. Cette particularité tire son origine du lien existant entre la protéine huntingtine et le récepteur HER2. Placées à la surface de la cellule, les protéines HER2 agissent comme des « interrupteurs » et maintiennent l’équilibre entre multiplication, division et réparation cellulaires. À l’inverse, dans certaines cellules tumorales, ces « interrupteurs » sont en nombre trop important avec pour conséquence une multiplication anarchique des cellules, ce qui joue un rôle capital sur le pronostic et le traitement de la maladie.
« Notre travail établit que la protéine huntingtine mutante interfère avec le bon fonctionnement du récepteur HER2, provoquant ainsi son accumulation au niveau de la membrane. Cette accumulation active des voies de signalisation induisant des métastases », précise Sandrine Humbert.
Pour arriver à cette conclusion, l’équipe de chercheurs, en collaboration avec des médecins de l’Institut Curie et de l’Hôpital de la Pitié-Salpêtrière, a travaillé à la fois sur des modèles de souris combinant l’affection neurodégénérative et un cancer du sein, et sur des cellules. L’approche cellulaire a permis de montrer l’effet de la huntingtine sur la sévérité du cancer en induisant une suractivation de la voie de signalisation HER2. La protéine huntingtine mutante perturbe la fonction d’une autre protéine, la dynamine, ce qui conduit à une accumulation du récepteur HER2 à la surface de la cellule.
Une protéine aux localisations multiples
La huntingtine est la protéine mutée responsable de la maladie de Huntington. Affection neurologique rare, cette maladie touche 1 personne sur 10 000 et se manifeste à l’âge adulte. Les symptômes les plus caractéristiques sont des troubles mentaux (anxiété, irritabilité, dépression), une détérioration intellectuelle qui progresse jusqu’à la démence, auxquels sont associés des mouvements anormaux involontaires et saccadés des membres, de la tête et du cou.


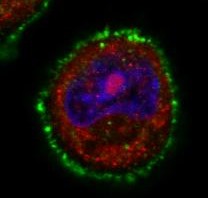
Dans cette cellule cancéreuse mammaire, on peut observer le récepteur Her2 (vert) et la dynamine localisée sous la membrane (rouge). Lorsque la huntingtine mutante (à droite) est exprimée, la dynamine présente une localisation plus diffuse, partiellement nucléaire (bleu) et la quantité d’Her2 à la membrane est augmentée. A gauche, cellule cancéreuse où la protéine huntingtine n’est pas mutée.
Barre d’échelle : 10 mM. © C. Moreira Sousa/Institut Curie
L’anomalie génétique qui provoque la maladie de Huntington est une augmentation anormale de la répétition de trois acides nucléiques (C, A et G – appelé triplet CAG) au sein du gène codant pour la protéine huntingtine. Normalement, ce triplet CAG se répète entre 17 et 35 fois contre 40 fois au moins dans le cas de la maladie de Huntington. Plus les répétitions sont nombreuses, plus les symptômes apparaissent tôt.
Cependant, l’expression de la huntingtine n’est pas limitée aux cellules nerveuses différenciées. Elle est également exprimée à des niveaux élevés dans d’autres types cellulaires, comme dans les cellules épithéliales d’origine non-neuronale. Des équipes de recherche commencent ainsi à s’intéresser aux manifestations périphériques de la maladie de Huntington. Chef d’équipe dans une fondation de soins et recherche en cancérologie, Sandrine Humbert s’est naturellement dirigée vers l’étude du rôle de la huntingtine dans le tissu mammaire. C’est ainsi que son équipe montre que plus les répétitions sont nombreuses dans la protéine huntingtine, plus le cancer apparaît précocement. « Examiner la fonction de cette protéine dans d’autres types cellulaires que les neurones permet d’approfondir nos connaissances sur la biologie de la cellule et par extension sur les mécanismes de dérégulation à l’origine du cancer et des maladies neurodégénératives» précise -t-elle.
Vers un meilleur suivi des patientes atteintes de la maladie de Huntington
« Grâce aux nouvelles données acquises, nous allons examiner de manière plus précise les tumeurs de patientes atteintes de la maladie de Huntington. Nous pourrons ainsi déterminer si effectivement ces dernières doivent bénéficier d’un suivi particulier. Nous avons démontré que la taille de l’expansion du triplet CAG dans le gène codant la protéine huntingtine, à l’origine de son caractère mutant, était corrélée à une apparition plus précoce du cancer. Pour autant nous ne pouvons conclure à une plus forte incidence de la survenue de cancers du sein chez les femmes atteintes de la maladie de Huntington. Des études cliniques récentes suggèrent même le contraire. Ainsi, l’incidence du cancer du sein pourrait être plus faible chez les patientes atteintes de la maladie de Huntington. Par contre, lorsqu’un cancer du sein se déclare, il pourrait être, dans certains cas spécifiques, plus agressif » explique Sandrine Humbert. En parallèle, les chercheurs souhaitent étudier le rôle de la protéine huntingtine normale dans le cancer du sein.