© AdobeStock
© AdobeStock
Des chercheurs de l’Inserm et de l’Université Évry Paris-Saclay au sein d’I-Stem, l’un des leaders européens de la recherche dans le domaine des cellules souches, ont développé une approche innovante cartographiant l’ensemble des effets biologiques des principes actifs de certains médicaments déjà sur le marché. Les premiers résultats, publiés dans Stem Cell Reports, ont permis d’identifier que la prazosine, une molécule utilisée normalement dans l’hypertension artérielle, pouvait avoir des effets thérapeutiques dans la sclérose latérale amyotrophique (SLA) et la dégénérescence fronto-temporale (FTD), deux maladies neuro-dégénératives liées à une insuffisance d’un gène essentiel au bon fonctionnement du nettoyage cellulaire.
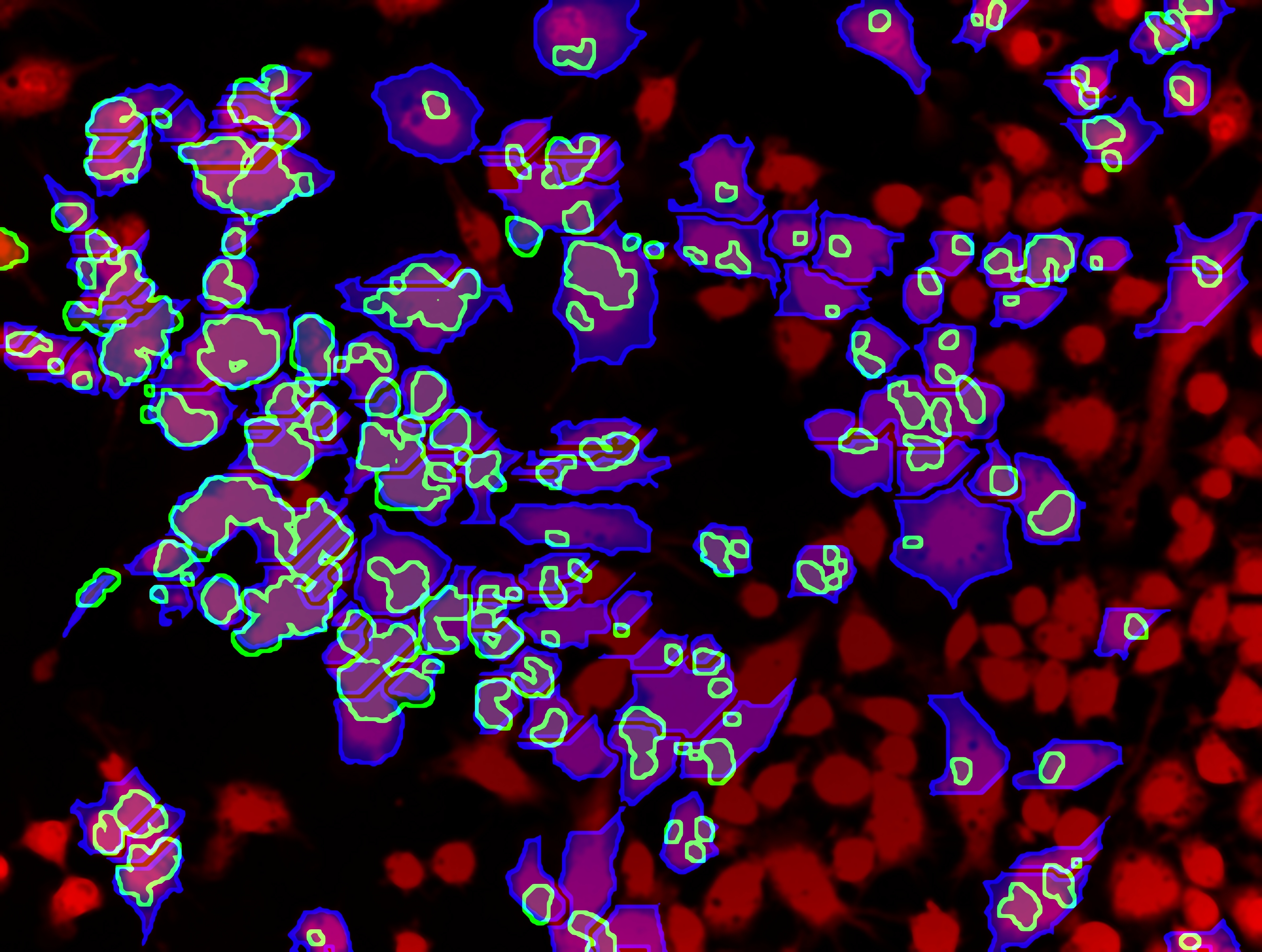
Le groupe de Sandrine Baghdoyan, ingénieure Inserm, au sein de l’unité UMR861 (Inserm/Université Évry Paris-Saclay), dirigée par Cécile Martinat, directrice de recherche Inserm, a élaboré une méthode innovante appelée « pharmacologie inverse » afin d’améliorer des techniques dites de criblage pharmacologique. L’approche traditionnelle par criblage consiste à évaluer les effets thérapeutiques potentiels de certaines molécules sur des modèles cellulaires de maladies ; une méthode efficace, mais souvent très longue. A contrario, la pharmacologie inverse consiste d’abord à établir une cartographie des effets biologiques de médicaments déjà sur le marché (effets sur l’expression des gènes, sur le fonctionnement des cellules, réponse immunitaire…) puis confronter cette signature aux mécanismes observés dans une maladie afin d’identifier les molécules les plus susceptibles d’avoir un effet thérapeutique
Les chercheuses et chercheurs[1] ont sélectionné 50 médicaments ayant un large spectre d’action et très peu d’effets secondaires afin de les tester sur des cellules saines et de déterminer et comparer leurs effets sur l’expression de l’ensemble des gènes. Les scientifiques ont ainsi montré que plus de la moitié des composés modifiaient significativement l’activité des gènes, dont plusieurs sont responsables de maladies.
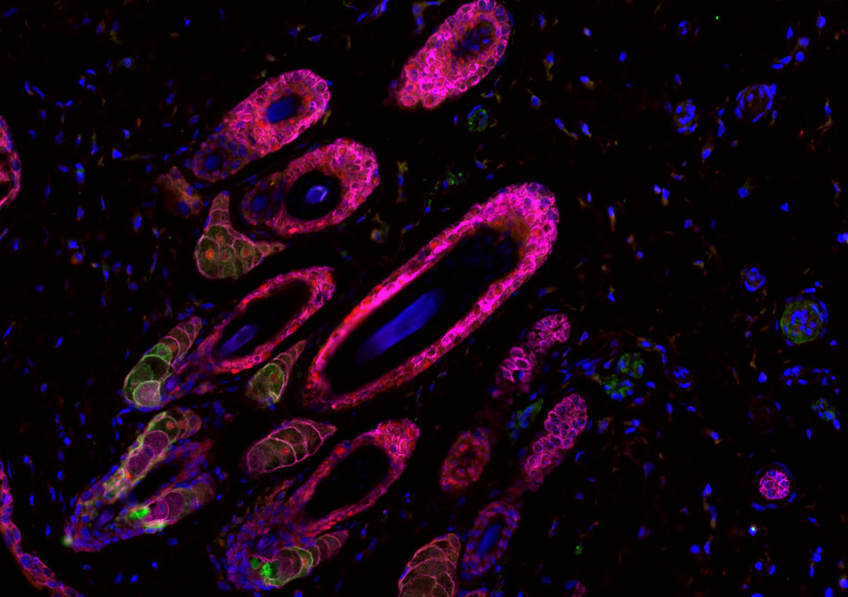
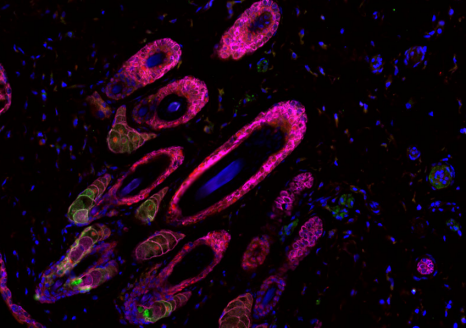
Cette stratégie innovante a permis d’identifier que la prazosine, un médicament déjà autorisé pour l’hypertension artérielle, pourrait avoir un effet thérapeutique dans deux maladies neurodégénératives : la sclérose latérale amyotrophique (SLA) et la dégénérescence fronto-temporale (FTD). Dans certaines formes de ces maladies, des mutations du gène SQSTM1 entrainent une diminution de l’expression de la protéine du même nom. Celle-ci joue un rôle clé dans l’autophagie, le mécanisme par lequel les cellules éliminent et recyclent leurs composants défectueux ou obsolètes. Les chercheurs ont montré que la prazosine augmentait l’expression de la protéine SQSTM1 et réactivait plusieurs voies moléculaires impliquées dans l’autophagie, suggérant une restauration de ce mécanisme essentiel au maintien de la santé des cellules.
Ces résultats ont ensuite été validés dans plusieurs modèles de SLA et de FTD, incluant des fibroblastes (cellules de soutien) issus de patients, des motoneurones dérivés de cellules souches pluripotentes humaines et un modèle de poisson-zèbre déficient en sqstm1.
« Nos travaux ont montré que la prazosine augmentait fortement l’expression du gène SQSTM1 déficient dans ces pathologies. Ces résultats sont encourageants et ouvrent la perspective d’un repositionnement de cette molécule pour ces maladies ultrares. Parallèlement, nous souhaitons établir le « profil » d’un plus grand nombre de molécules afin de disposer d’une sorte de bibliothèque pour étendre la pharmacologie inverse à d’autres maladies » précise Cécile Martinat.
[1] Ces travaux ont également été réalisés en collaboration avec le Centre National de Recherche en Génomique Humaine (CNRGH), l’École normale supérieure de Lyon, l’Institut Imagine et l’Institut du Cerveau.







 © Photo de
© Photo de