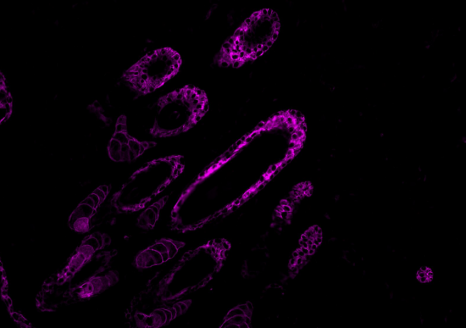
Co-marquages de peau de souris exprimant une mutation du gène PIK3CA. ©Marina Firpion/Guillaume Canaud – unité 1151 Inserm
Trois millions de Français sont concernés par les maladies rares et près de 80% de ces maladies sont d’origine génétique. Etudier les mécanismes impliqués dans ces pathologies représente un enjeu de taille pour la recherche : si cela permet avant tout d’aider les patients, les découvertes réalisées peuvent permettre de décrypter des mécanismes plus généraux, relatifs à d’autres maladies. A l’approche de la Journée Internationale des maladies rares le 28 février 2022, l’Inserm fait le point sur 3 récentes études témoignant des avancées de la recherche sur certaines de ces maladies rares.
De nouvelles perspectives thérapeutiques pour les nourrissons atteints de syndromes d’hypercroissance dysharmonieuse
Les syndromes d’hypercroissance dysharmonieuse sont des maladies génétiques rares caractérisées par une augmentation de la taille mais aussi du nombre de cellules dans le corps. Ils se manifestent par une asymétrie pouvant toucher n’importe quelle partie ou tissu du corps (graisse, vaisseaux, muscles, os…), y compris le cerveau. Dans 95 % des cas, la maladie est liée à une mutation, lors du développement embryonnaire, du gène PIK3CA, qui régule la prolifération et la croissance des cellules.
Depuis 2016, une équipe composée de chercheurs de l’Inserm, de l’AP-HP, d’Université de Paris, à l’Institut Necker-Enfants malades et des services cliniques des Hospices civils de Lyon a démontré l’efficacité thérapeutique d’une molécule utilisée contre certains cancers, l’Alpelisib, un médicament inhibiteur de PIK3CA, pour traiter un groupe d’enfants et d’adultes présentant des formes sévères de ces maladies. Dans une récente publication dans le Journal of Experimental Medicine, l’équipe décrit une amélioration à la fois clinique, biologique et d’imagerie chez deux nourrissons atteints de formes sévères de syndromes d’hypercroissance dysharmonieuse et traités par Alpelisib. Il s’agit des premières données sur les effets de cette molécule obtenues dans des formes néonatales graves.
Lire le communiqué de presse du 26/01/22 : Amélioration de la santé de deux nourrissons atteints de formes sévères de syndromes d’hypercroissance dysharmonieuse
Identification d’une cause génétique d’une forme rare du syndrome de Cushing induit par l’alimentation
L’hyperplasie bilatérale macronodulaire des surrénales avec syndrome de Cushing induit par l’alimentation est une maladie rare qui touche les deux glandes surrénales situées au-dessus des reins et entraine une surproduction de cortisol, une hormone stéroïde dont l’excès a des conséquences néfastes pour l’organisme.
Dans une étude récemment publiée dans la revue The Lancet Diabetes & Endocrinology, une équipe de chercheurs et chercheuses du service d’endocrinologie et des maladies de la reproduction de l’hôpital Bicêtre AP-HP, de l’Inserm et de l’Université Paris-Saclay, est parvenue à identifier les déterminants moléculaires de la survenue de cette maladie 30 ans après sa description initiale. Ils ont mis à jour les mutations responsables de cette forme du syndrome de Cushing dans le gène KDM1A
Lire le communiqué de presse du 05/11/21 : Une étude de cohorte permet d’identifier une cause génétique d’une forme rare du syndrome de Cushing induit par l’alimentation
Découverte d’une première cause génétique pour le syndrome de l’homme-arbre
Une grande partie de la population est porteuse de papillomavirus humains (HPVs), et notamment de papillomavirus cutanés, qui provoquent en général des verrues ou des lésions locales et bénignes. Toutefois de très rares patients dans le monde développent des formes sévères de ces maladies virales, dont le syndrome de « l’homme-arbre ». Cette maladie très handicapante se manifeste par une poussée anarchique de cornes cutanées, pour lesquelles la chirurgie n’est pas efficace.
Dans le cadre d’une collaboration internationale, des chercheurs de l’Inserm et enseignants-chercheurs d’Université de Paris et médecins de l’AP-HP regroupés à l’Institut Imagine (Inserm/Université de Paris, AP-HP) situé au sein de l’hôpital Necker-Enfants malades AP-HP ont pour la première fois mis en évidence une cause génétique de ce syndrome : une mutation du gène CD28. Ces résultats ont été publiés dans la revue Cell.
Lire le communiqué de presse du 02/07/21 : Découverte d’une première cause génétique pour le syndrome de l’homme-arbre (papillomavirus cutané)
Ces contenus pourraient aussi vous intéresser :

Contacts