Brain scan, X-ray © Fotolia
Dysfunctions of ion channels – or channelopathies – in the brain are today associated with more than 30 neurological diseases such as epilepsy or cerebellar ataxias. Structures located on the membrane of cells allowing the passage of ions (for example sodium and potassium ions) between the interior of a cell and its external environment (extracellular environment), these channels make it possible in particular to generate and control d potentials. action in neurons. A study conducted at the Brain Institute (Sorbonne University / Inserm / AP-HP / CNRS) identified a new cerebral channelopathy originating from dominant mutations in the KCNN2 gene, encoding the SK2 ion channel. The results were published in Brain on November 27, 2020.
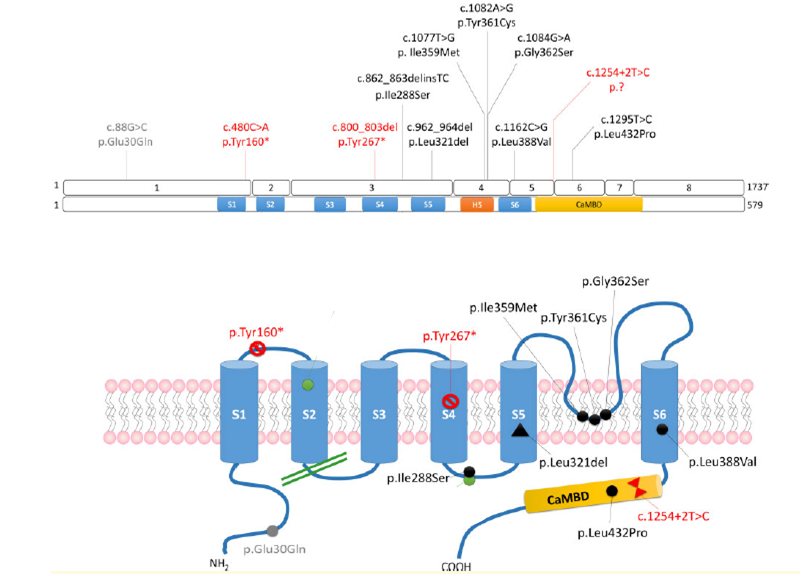
Pathogenic variants of the KCNN2 gene identified in patients and their location on the protein structure of the SK2 channel.
The variants in red are pathogenic variants truncating (introducing a stop codon into the protein sequence). Variants in black are pathogenic missense variants associated with loss of function. The variant in gray was classified of unknown significance because the channel with this variant did not show any particular deficit in electrophysiology.
Dr Fanny Mochel, geneticist in the genetics department of the Pitié-Salpêtrière hospital AP-HP and researcher at the Brain Institute (Sorbonne University / Inserm / AP-HP / CNRS) and Professor Christel Depienne, A geneticist at the Institute of Human Genetics at the University Hospital of Essen (Germany) and also a researcher at the Brain Institute have identified a new syndrome associated with mutations in the SK2 channel. The study published in the scientific journal Brainconcerns 10 patients, 6 men and 4 women aged 2 to 60 years with more or less severe intellectual delays associated, for some, with autism spectrum disorders or psychotic episodes. These cognitive disorders are in all cases associated with tremors, symptoms of cerebellar ataxia or even abnormal movements.
Thanks to a collaboration with Agnes Rastetter from the genotyping / sequencing platform of the Brain Institute (Sorbonne University / Inserm / AP-HP / CNRS), the genome of a first patient recruited at Pitié-Salpêtrière was analyzed at the search for genetic mutations at the origin of this syndrome. This analysis revealed a mutation in the KCNN2 gene interrupting its coding sequence, absent from the patient’s parents ( de novo mutation ). Brain imaging by MRI (magnetic resonance imaging) in this patient showed abnormalities in the structure and integrity of the white matter of the brain, that is, the cerebral sheath that protects the axons of neurons.
In addition, an international collaboration has enabled researchers to identify 9 other patients with mutations in the KCNN2 gene . The majority of these mutations had arisen de novo while a mutation was transmitted in a familial form of the same syndrome.
Finally, by working jointly with Carine Dalle from the electrophysiology cell exploration platform of the Brain Institute, the teams of Dr Mochel and Depienne have shown a deleterious role of these mutations on the function of the SK2 channel, i.e. that is to say a loss of function leading to a dysfunction of the ion channel SK2 and therefore a loss of regulation of the action potential, support of the nervous message.
The results of this new study have identified a new cerebral channelopathy originating from dominant mutations in the KCNN2 gene , encoding the SK2 ion channel. This new syndrome is characterized by the presence, on the one hand, of cognitive symptoms, in particular an intellectual disability, and, on the other hand, of motor symptoms such as abnormal movements.
This new pathology, the cause of which is now known, is very heterogeneous from a point of view of symptoms and requires multidisciplinary management at the border between genetics, for the search for mutations in the KCNN2 gene, pediatric neurology and neurology. for the management of cognitive and motor manifestations of patients.
These contents could be interesting :