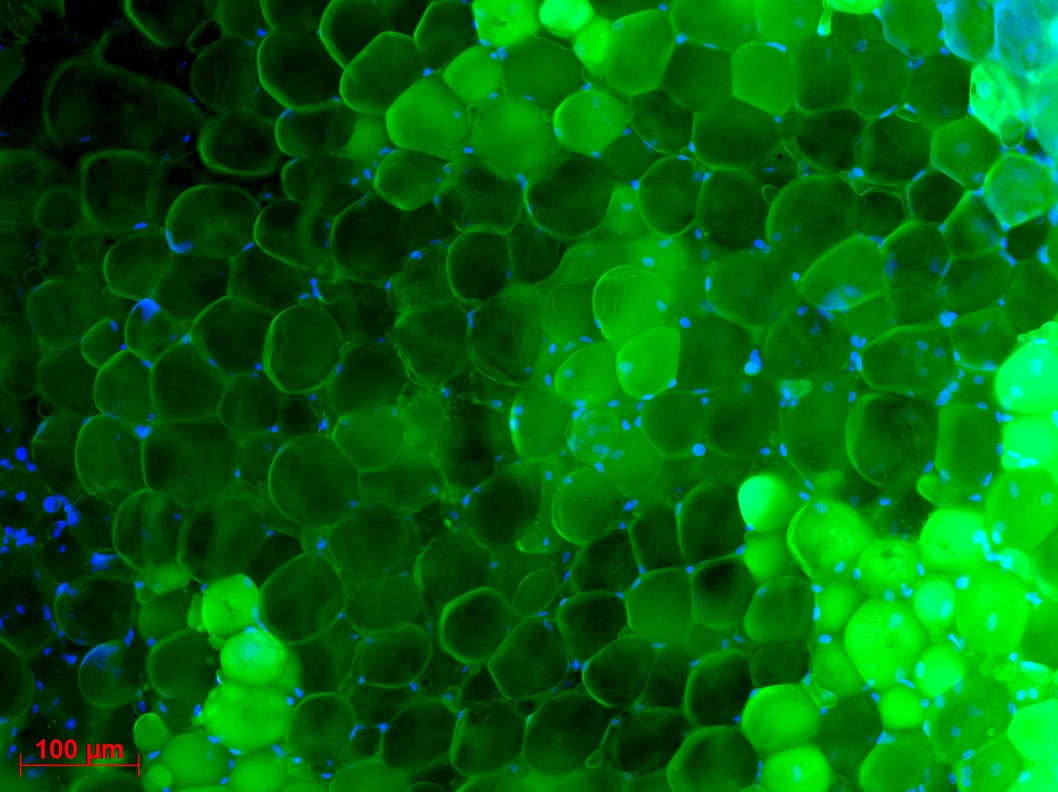
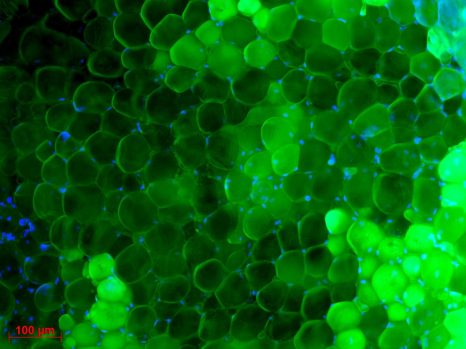
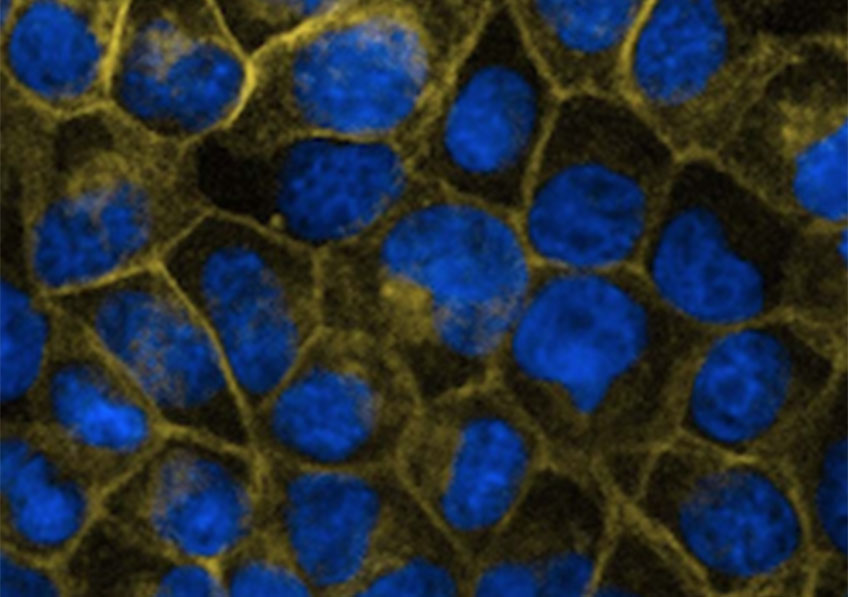
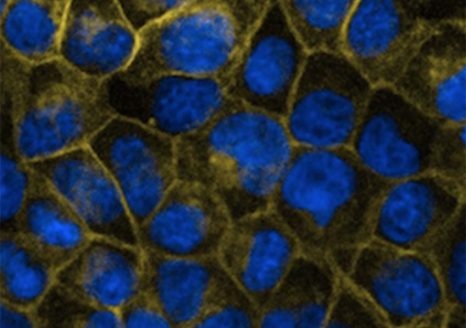
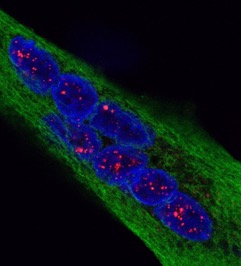
Fibroblastes humains observés en microscopie de fluorescence. Les mitochondries sont marquées en rouge, l’ADN du noyau des fibroblastes est marqué en bleu. © Nivea Dias Amoedo/Inserm
Fibroblastes humains observés en microscopie de fluorescence. Les mitochondries sont marquées en rouge, l’ADN du noyau des fibroblastes est marqué en bleu. © Nivea Dias Amoedo/Inserm
Une dégradation de la qualité de la peau, de sa capacité à cicatriser, et de son vieillissement normal, est souvent observée chez les personnes présentant une hyperglycémie chronique. Une équipe de chercheuses et chercheurs de l’Inserm, de l’université de Bordeaux et de LVMH Recherche, s’est intéressée à la façon dont l’hyperglycémie altère le derme humain et en particulier les cellules impliquées dans sa cicatrisation, les fibroblastes. Ses travaux, parus dans Redox Biology, montrent qu’une trop forte concentration en glucose dans le derme vient perturber une mécanique complexe et finement régulée de production de l’énergie par les fibroblastes, avec des impacts sur leur capacité à maintenir l’intégrité de la peau.
Le glucose est un sucre vital pour les cellules des mammifères : il permet notamment la synthèse de nombreuses molécules essentielles à l’organisme, comme l’ADN, ainsi que la transformation d’énergie par les mitochondries, les « centrales énergétiques » du corps humain, via le mécanisme dit de « respiration mitochondriale ». Bien que les concentrations en glucose dans le derme (l’une des trois couches constituant la peau, située entre l’épiderme – la couche externe – et l’hypoderme) reflètent celles retrouvées dans le sang, le métabolisme du glucose dans la peau reste peu étudié et mal connu.
Au sein du derme, on retrouve les fibroblastes, des cellules, impliquées notamment dans la régénération de l’épiderme et dans la cicatrisation de la peau, grâce à leur capacité à produire du collagène et à se déplacer sur le site d’une blessure. Ces fibroblastes cutanés subissent directement le stress métabolique causé par l’hyperglycémie[1], une conséquence des régimes alimentaires riches en sucres.
Or, l’hyperglycémie et les maladies métaboliques qui lui sont liées (comme le diabète par exemple) sont fréquemment associées à une dégradation de la qualité et de l’intégrité de la peau, avec en particulier une moins bonne cicatrisation et un vieillissement cutané prématuré. Une des clés pour limiter ces altérations pourrait ainsi être de mieux comprendre comment l’hyperglycémie impacte le métabolisme et la structure de la peau.
Une équipe projet co-dirigée par Rodrigue Rossignol, directeur de recherche Inserm et co-directeur du laboratoire Maladies rares : génétique et métabolisme (Inserm/université de Bordeaux) et Anne-Laure Bulteau à LVMH Recherche, s’est intéressée à la façon dont les fibroblastes du derme humain et les mitochondries qu’ils contiennent se comportent lorsqu’ils sont exposés à plusieurs degrés d’hyperglycémie : normale, modérée, élevée et extrême[2].
Ces études ont été menées dans 4 modèles complémentaires : sur des fibroblastes cultivés in vitro, dans un derme reconstitué (un modèle in vitro reproduisant la structure en trois dimensions du derme), dans une peau humaine reconstituée (similaire au derme reconstitué mais combinant derme et épiderme) et enfin dans une biopsie cutanée prélevée sur un patient diabétique.
Les résultats mettent en évidence un système très sensible et complexe de régulation du métabolisme énergétique et de l’activité des mitochondries au sein des fibroblastes humains, en réponse à la variation du taux de glucose dans le derme.
Les scientifiques ont notamment constaté qu’une hyperglycémie croissante inhibe la respiration cutanée réalisée par les mitochondries. Ils ont identifié des mécanismes moléculaires inédits débutant par la répression de l’activité des mitochondries, puis menant à leur fragmentation, jusqu’à l’activation de leur dégradation.
« Le blocage de la chaîne respiratoire des mitochondries produit des molécules toxiques pour la peau, impliquées dans son vieillissement, explique Rodrigue Rossignol, on parle alors de stress oxydatif. »
Parmi les acteurs de la régulation de l’activité mitochondriale, les scientifiques ont identifié un facteur de croissance, appelé GDF15, dont l’activité était fortement inhibée dès l’apparition de l’hyperglycémie modérée et qui tendait à continuer de diminuer avec l’augmentation du taux de glucose environnant. Cette inhibition entraînait alors la diminution de la production de nouvelles mitochondries dans les fibroblastes. En revanche, une supplémentation des modèles de peau en GDF15 permettait d’inverser les altérations observées du métabolisme énergétique, même si l’hyperglycémie persistait.
« Nos résultats suggèrent que GDF15 pourrait être au cœur d’une potentielle stratégie pharmacologique ou dermatologique visant à limiter les dommages cutanés causés par le stress métabolique chez les personnes hyperglycémiques, indique Rodrigue Rossignol. Le chercheur tempère cependant : en conditions réelles, l’hyperglycémie chronique implique des phénomènes inflammatoires. Ceux-ci, non reproduits dans nos modèles, pourraient être susceptibles d’entraver l’action protectrice d’une supplémentation en GDF15. »
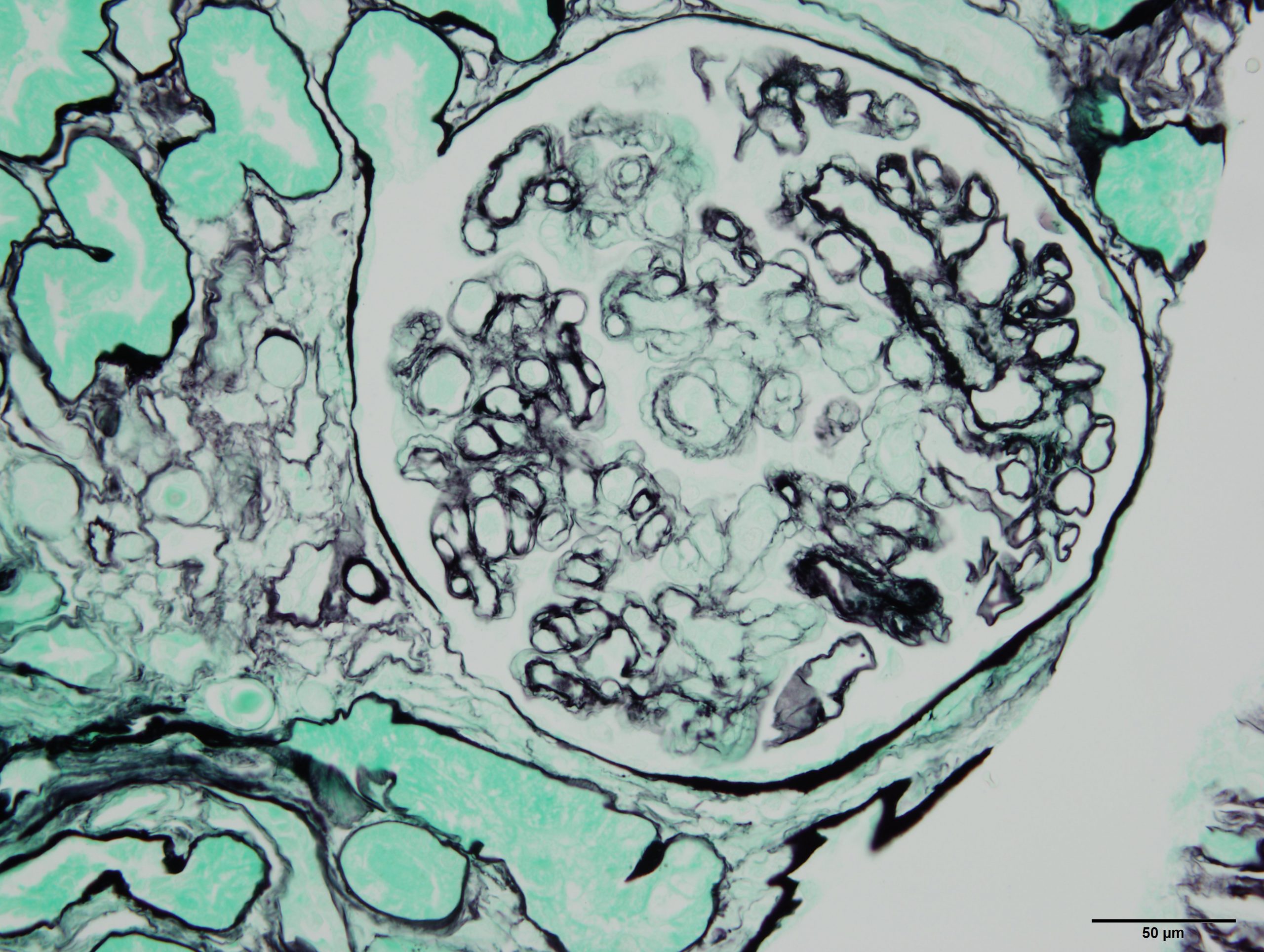
Enfin, l’équipe a pu observer que l’altération de l’activité mitochondriale des fibroblastes dégradaient leur capacité à produire un réseau de collagène cutané qualitatif.
« En cas de lésion cutanée, le réseau de collagène sert notamment aux fibroblastes à se déplacer dans le derme pour aller réparer les zones abîmées, détaille Rodrigue Rossignol, nos résultats montrent que, sous l’effet de l’hyperglycémie, le réseau étant défaillant, les fibroblastes s’y déplaçaient plus difficilement et la reconstruction cutanée était donc moins efficace. »
Ces données apportent de nouvelles connaissances fondamentales sur la façon dont l’hyperglycémie altère la physiologie de la peau et des mitochondries. Elles offrent de nouvelles perspectives concernant les causes de la dégradation de la qualité de la peau chez les personnes présentant une hyperglycémie et ouvrent la voie à de potentielles stratégies innovantes ciblant spécifiquement les mitochondries
Ces travaux sont co-financés par l’Inserm, l’université de Bordeaux, LVMH Recherche, la Fondation pour la recherche médicale (FRM) et la région Nouvelle-Aquitaine.
Seyta Ley Ngardigal, première autrice de cette étude et docteure de l’université de Bordeaux, a bénéficié d’une bourse doctorale financée par LVMH Recherche. Ces travaux s’inscrivent ainsi dans le cadre d’une thèse CIFRE dirigée par Rodrigue Rossignol et Anne-Laure Bulteau, au sein de l’unité Maladies Rares : génétique et métabolisme (Inserm/université de Bordeaux) et de LVMH Recherche (Orléans).
Les CIFRE, ou Conventions industrielles de formation par la recherche, sont des dispositifs financés par le ministère chargé de l’Enseignement supérieur de la Recherche ayant pour vocation à renforcer les échanges entre les laboratoires de recherche publique et les milieux socio-économiques, favoriser l’emploi des docteurs dans les entreprises et contribuer au processus d’innovation des entreprises établies en France.
[1]Selon l’OMS, un niveau de concentration sanguine en glucose normal est de 700 à 1000 mg de glucose par litre de sang (3,9 à 5,6 mmol/L). Entre 1200 et 2162mg/L (6,9 à 12 mmol/L), la personne est considérée en hyperglycémie ; 1200mg/L est considérée comme une hyperglycémie modérée et 2162 mg/L comme une hyperglycémie élevée.
[2]25mmol/L, soit environ deux fois la valeur d’une hyperglycémie élevée.