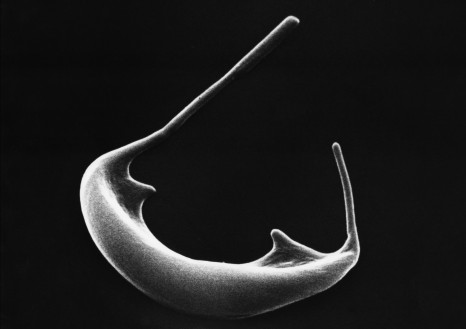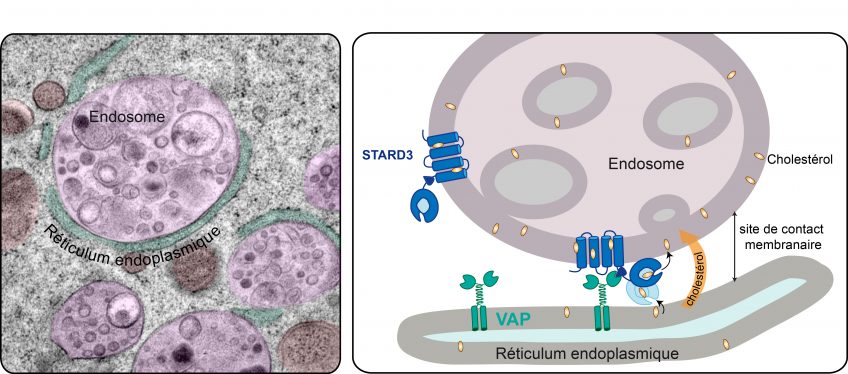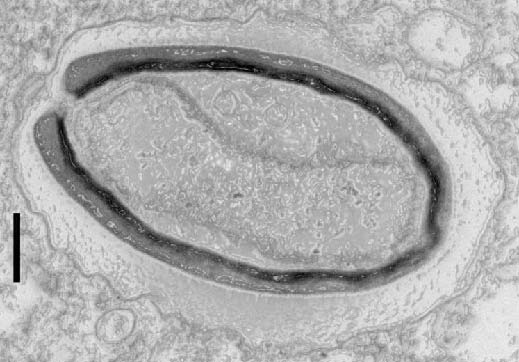
Pandoravirus quercus, found in Marseille, France. Thin section, viewed via electron microscopy. Scale bar: 100 nm. ©IGS- CNRS/AMU.
Three new members have been isolated and added to the Pandoravirus family by researchers at the Structural and Genomic Information Laboratory (CNRS/Aix‐Marseille Université), working with partners at the Large Scale Biology Laboratory (CEA/Inserm/Université Grenoble‐Alpes) and at CEA-Genoscope. This strange family of viruses, with their giant genomes and many genes with no known equivalents, surprised the scientists when they were discovered a few years ago. In the 11 June 2018 edition of Nature Communications, researchers offer an explanation: pandoviruses appear to be factories for new genes – and therefore new functions. From freaks of nature to evolutionary innovators, giant viruses continue to shake branches on the tree of life!
In 2013, the discovery of two giant viruses unlike anything seen before blurred the line between the viral and cellular world. Pandoraviruses are as big as bacteria, and contain genomes that are more complex than those found in some eukaryotic organisms[1]. Their strange amphora shape and enormous, atypical genome[2] led scientists to wonder where they came from.
The same team has since isolated three new members of the family in Marseille, continental France, Nouméa, New Caledonia, and Melbourne, Australia. With another virus found in Germany, the team compared those six known cases using different approaches. Analyses showed that despite having very similar shapes and functions, these viruses only share half of their genes coding for proteins. Usually, however, members of the same family have more genes in common.
Furthermore, these new members contain a large number of orphan genes, i.e. genes which encode proteins that have no equivalent in other living organisms (this was already the case for the two previously discovered pandoraviruses). This unexplained characteristic is at the heart of many a debate over the origin of viruses. What most surprised researchers was that the orphan genes differed from one pandoravirus to another, making it less and less likely that they were inherited from a common ancestor!
Bioinformatic analysis showed that these orphan genes exhibit features very similar to those of non-coding (or intergenic) regions in the pandoravirus genome. Findings indicate the only possible explanation for the gigantic size of pandoravirus genomes, their diversity and the large proportion of orphan genes they contain: most of these viruses’ genes may originate spontaneously and randomly in intergenic regions. In this scenario, genes “appear” in different locations from one strain to another, thus explaining their unique nature.
If confirmed, this groundbreaking hypothesis would make these giant viruses craftsmen of genetic creativity – a central, but still poorly explained component of any understanding of the origin of life and its evolution.
[1] Organisms whose cells contain nuclei, unlike the two other kingdoms of living organisms, bacteria and archaea.
[2] Up to 2.7 million base pairs.
Credit: IGS- CNRS/AMU