©Inserm/Cochet-Escartin, Olivier, 2014
Asymmetry plays a major role in biology at every scale: think of DNA spirals, the fact that the human heart is positioned on the left, our preference to use our left or right hand … A team from the Institute of biology Valrose (CNRS/Inserm/Université Côte d’Azur), in collaboration with colleagues from the University of Pennsylvania, has shown how a single protein induces a spiral motion in another molecule. Through a domino effect, this causes cells, organs, and indeed the entire body to twist, triggering lateralized behaviour. This research is published in the journal Science on November 23, 2018.
Our world is fundamentally asymmetrical: think of the double helix of DNA, the asymmetrical division of stem cells, or the fact that the human heart is positioned on the left … But how do these asymmetries emerge, and are they linked to one another?
At the Institute of biology Valrose, the team led by the CNRS researcher Stéphane Noselli, which also includes Inserm and Université Cote d’Azur researchers, has been studying right–left asymmetry for several years in order to solve these enigmas. The biologists had identified the first gene controlling asymmetry in the common fruit fly (Drosophila), one of the biologists’ favoured model organisms. More recently, the team showed that this gene plays the same role in vertebrates: the protein that it produces, Myosin 1D,[1] controls the coiling or rotation of organs in the same direction.
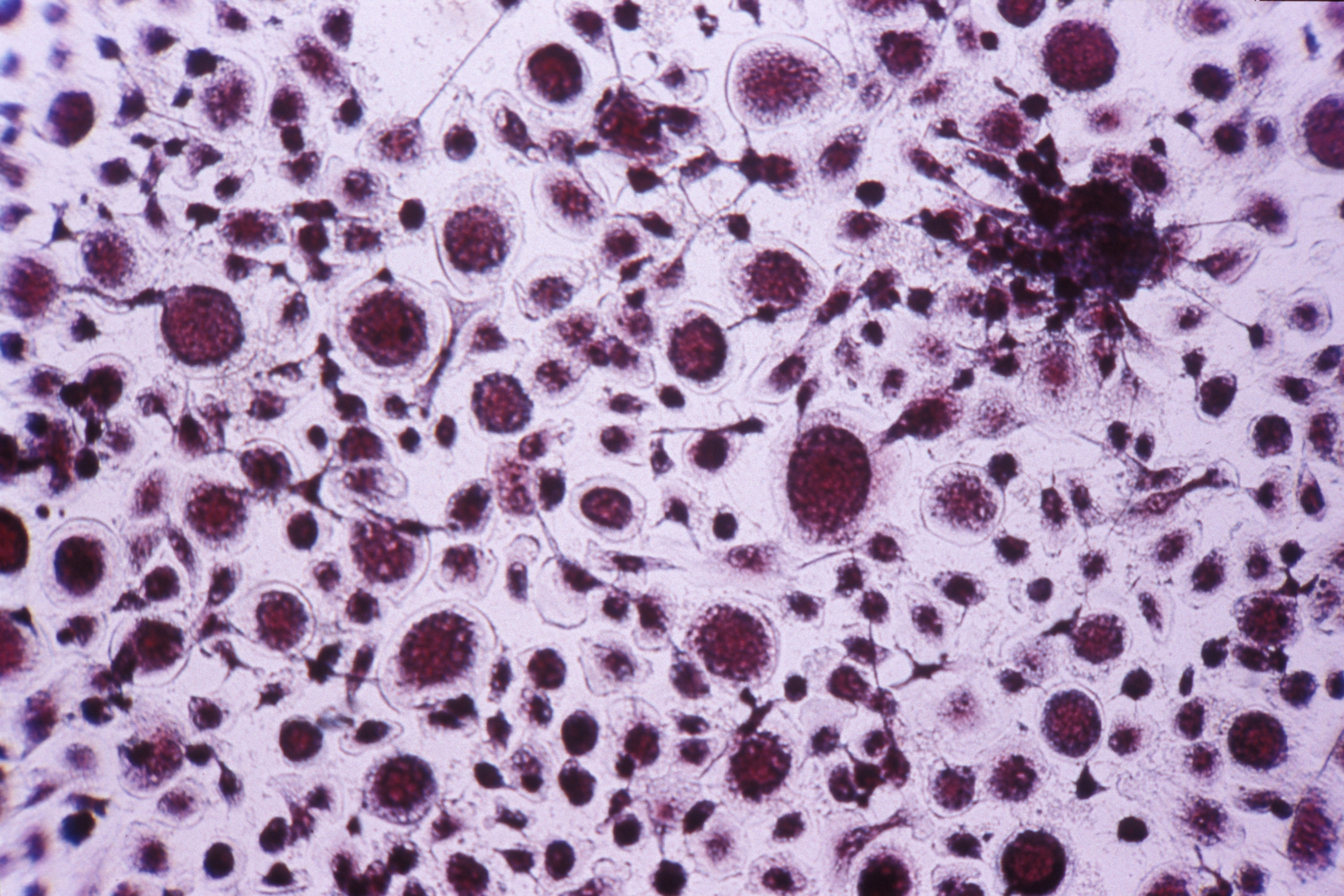
In this new study, the researchers induced the production of Myosin 1D in the normally symmetrical organs of Drosophila, such as the respiratory trachea. Quite spectacularly, this was enough to induce asymmetry at all levels: deformed cells, trachea coiling around themselves, the twisting of the whole body, and helicoidal locomotive behavior among fly larvae. Remarkably, these new asymmetries always develop in the same direction.
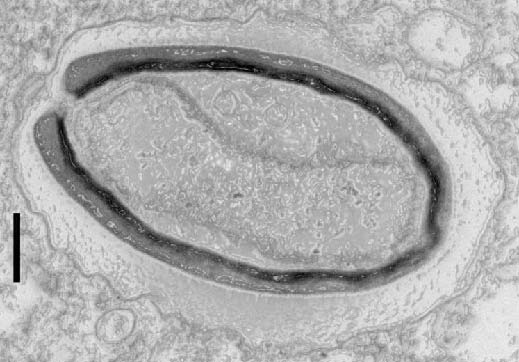
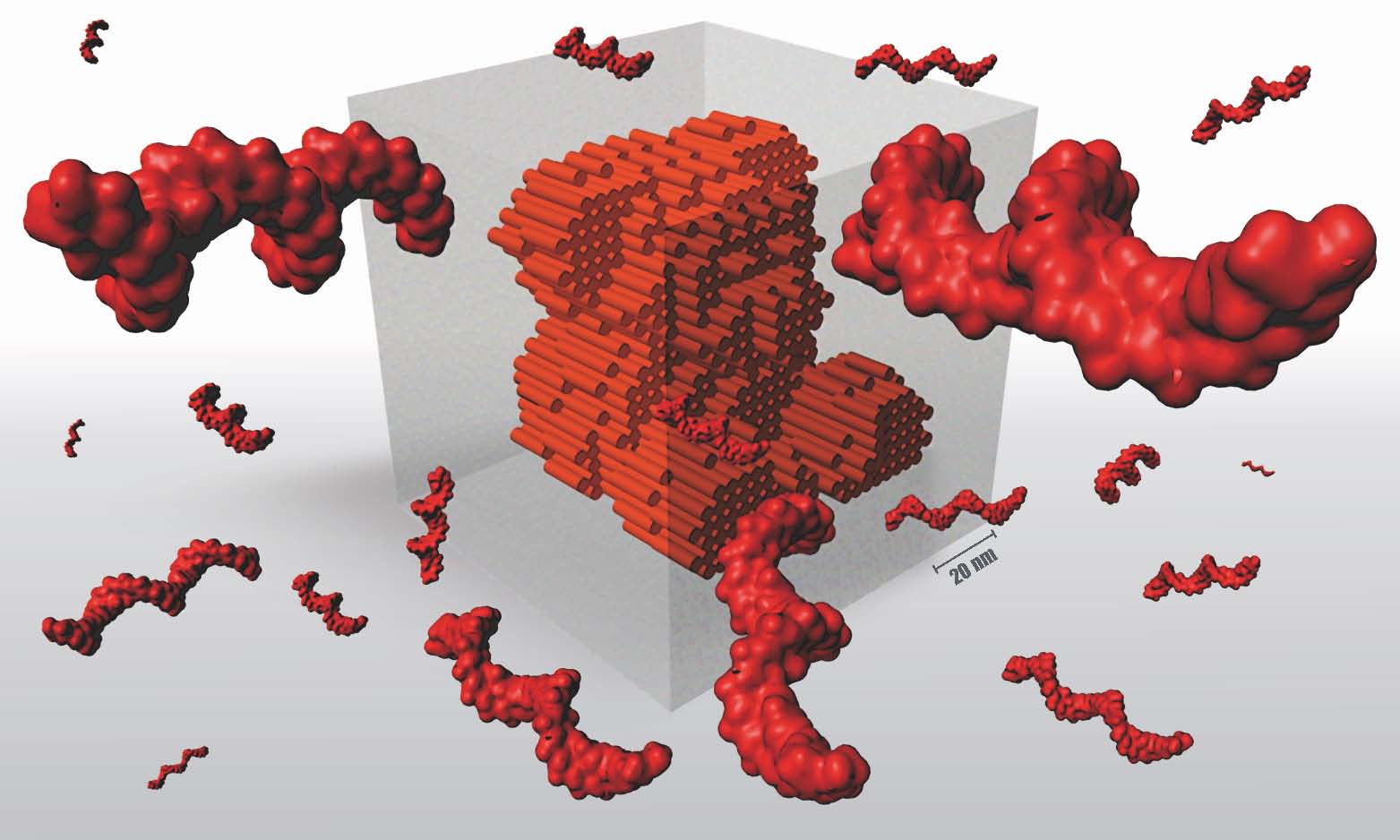
In order to identify the origin of these cascading effects, biochemists from the University of Pennsylvania contributed to the project too: on a glass coverslip, they brought Myosin 1D into contact with a component of cytoskeleton (the cell’s “backbone”), namely actin. They were able to observe that the interaction between the two proteins caused the actin to spiral.
Besides its role in right–left asymmetry among Drosophila and vertebrates, Myosin 1D appears to be a unique protein that is capable of inducing asymmetry in and of itself at all scales, first at the molecular level, then, through a domino effect, at the cell, tissue, and behavioral level.
These results suggest a possible mechanism for the sudden appearance of new morphological characteristics over the course of evolution, such as, for example, the twisting of snails’ bodies. Myosin 1D thus appears to have all the necessary characteristics for the emergence of this innovation, since its expression alone suffices to induce twisting at all scales.
[1] Myosins are a class of proteins that interact with actin (a constituent of cell skeletons or cytoskeletons). The most well-known of them, muscular myosin, makes muscles contract.