Human kidney section magnified 400 times by a polychromatic immunofluorescence microscope. © Inserm/Oriol, Rafael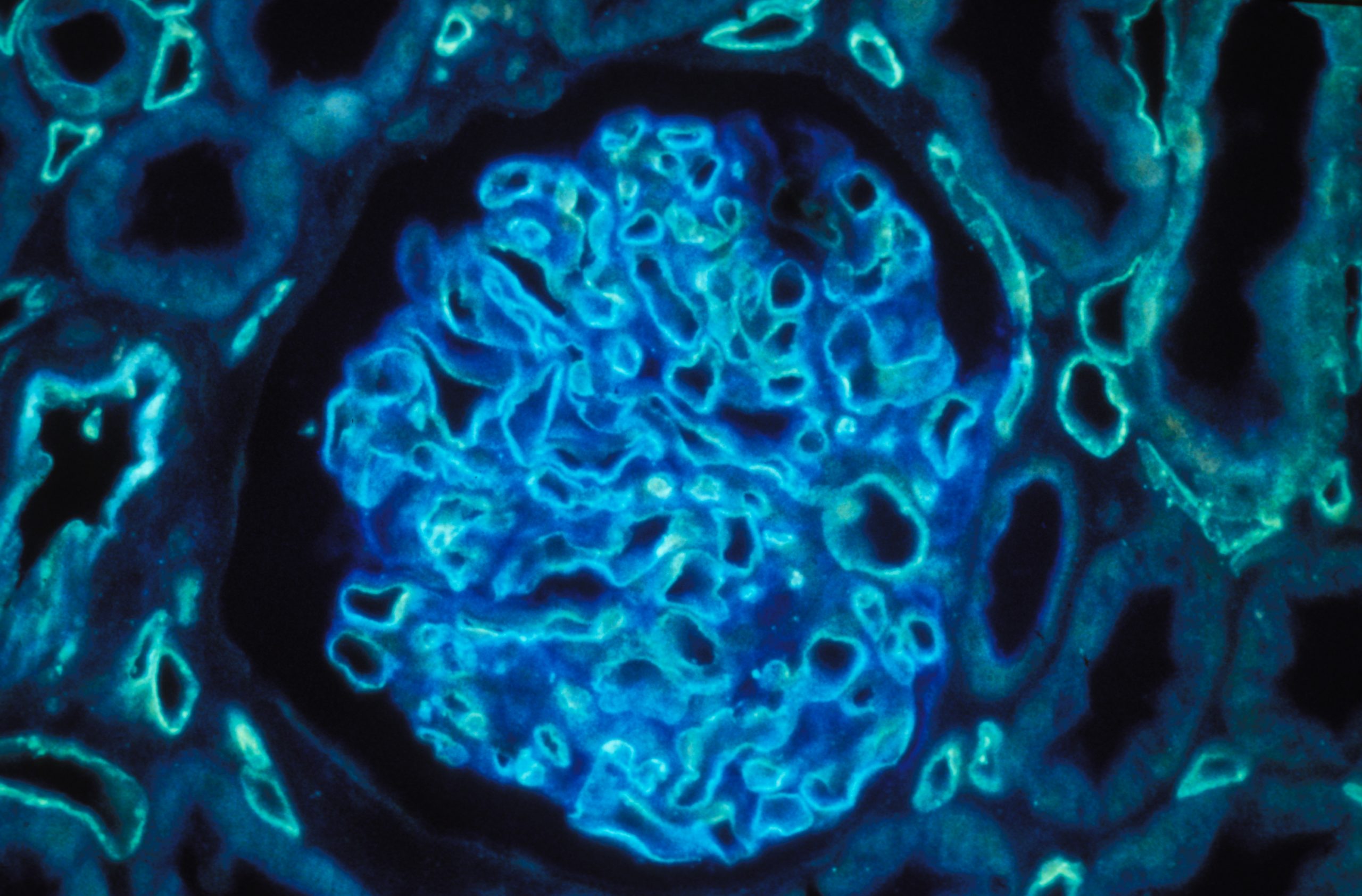
The team made up of researchers from the endocrinology and reproductive diseases department of Bicêtre AP-HP hospital, Inserm and Paris-Saclay University, carried out work, coordinated by Professor Peter Kamenický, to study the genetic cause of bilateral macronodular adrenal hyperplasia with diet-induced Cushing’s syndrome. This rare disease affects the two adrenal glands located above the kidneys and causes an overproduction of cortisol, a steroid hormone whose excess has harmful consequences for the body . Researchers were able to determine the molecular explanation for the occurrence of this disease 30 years after its initial description. This work was published on October 13, 2021 in the journal The Lancet Diabetes & Endocrinology .
This rare form of adrenal Cushing syndrome, studied by these researchers, is due to the abnormal expression of the GIP receptor (Glucose-dependent insulinotropic peptide) in both adrenal glands of patients. GIP is a hormone produced by the small intestine in response to the ingestion of food. In patients with this particular form of Cushing’s syndrome, cortisol levels increase abnormally after each food intake. Patients with this disease develop the typical clinical signs of Cushing’s syndrome such as weight gain associated with muscle atrophy, high blood pressure, diabetes mellitus, osteoporosis, and depression. The pathology is associated with an increase in mortality, especially cardiovascular causes.
In this international study involving researchers from six countries, and based in particular on close Franco-Quebec collaboration, the team reports that GIP-dependent macronodular hyperplasia of the adrenals, in both familial and sporadic forms, is a genetic disease caused by germline mutations of Lysine Demethylase 1A (KDM1A) with secondary loss of the second KDM1A locus, comprising the second copy of the gene, in adrenal tissue. KDM1A acts mainly as a transcriptional repressor (ie a regulator which prevents a gene from being expressed), the loss of its function results in deregulation of the expression of various genes in the adrenal tissue, including the GIP receptor but also of other receptors coupled to G proteins.
This discovery will make it possible to offer genetic counseling and earlier detection of this rare disease to patients and their relatives. Rare diseases are generally underdiagnosed. This is all the more important as the pathogenic variations of KDM1A also predispose to myeloma and other types of cancer.
In addition, this new role of KDM1A as an epigenetic regulator of the expression of the GIP receptor and other receptors coupled to G proteins could have pharmacological implications.
These contents could be interesting :

Medias
Sources
The Lancet Diabetes & Endocrinology.
Fanny Chasseloup, Isabelle Bourdeau, Antoine Tabarin, Daniela Regazzo, Charles Dumontet, Nataly Ladurelle, Lucie Tosca, Larbi Amazit, Alexis Proust, Raphael Scharfmann, Tiphaine Mignot, Frédéric Fiore, Stylianos Tsagarakis, Dimitra Vassiliadi, Dominique Maiter, Jacques Young, Anne-Lise Lecoq, Vianney Deméocq, Sylvie Salenave, Hervé Lefebvre, Lucie Cloix, Philippe Emy, Rachel Dessailloud, Delphine Vezzosi, Carla Scaroni, Mattia Barbot, Wouter de Herder, François Pattou, Martine Tétreault, Gilles Corbeil, Margot Dupeux, Benoit Lambert, Gérard Tachdjian, Anne Guiochon-Mantel, Isabelle Beau, Philippe Chanson, Say Viengchareun, André Lacroix, Jérôme Bouligand, Peter Kamenický.