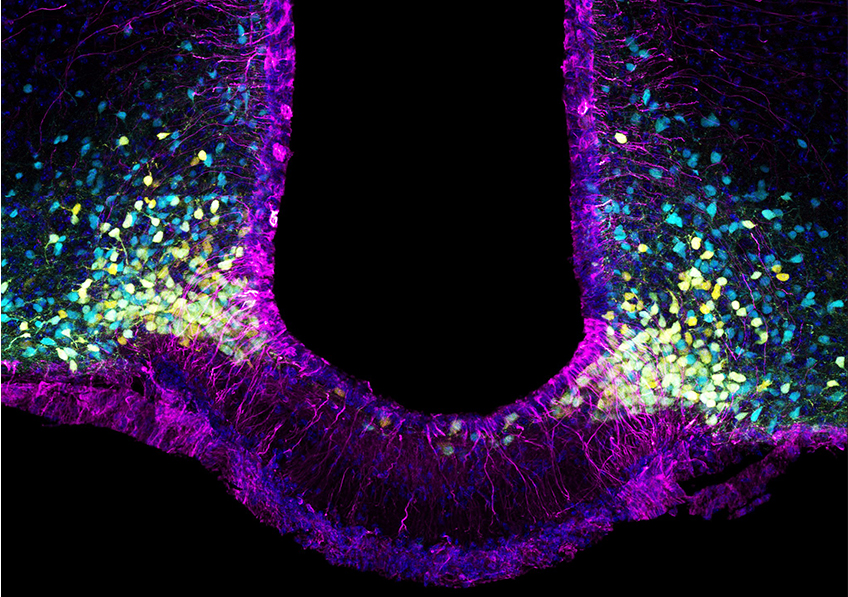
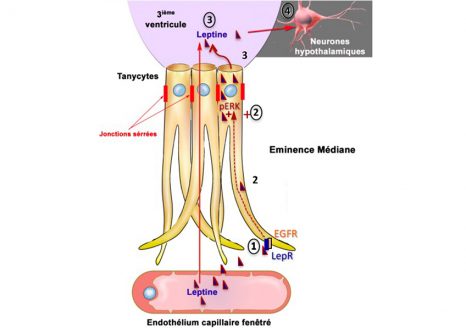
Leptin brain entry via a tanycytic LepR:EGFR shuttle controls lipid metabolism and pancreas function
Manon Duquenne1, Cintia Folgueira2#, Cyril Bourouh3#, Marion Millet4,#, Anisia Silva5#, Jérôme Clasadonte1#, Monica Imbernon1, Daniela Fernandois1, Ines Martinez-Corral1, Soumya Kusumakshi6, Emilie Caron1, S. Rasika1, Eleonora Deliglia1, Nathalie Jouy1,12, Asturo Oishi5, Massimiliano Mazzone7, Eric Trinquet8, Jan Tavernier9, Young-Bum Kim11, Stéphane Ory4, Ralf Jockers5, Markus Schwaninger10, Ulrich Boehm6, Ruben Nogueiras2, Jean-Sébastien Annicotte3, Stéphane Gasman4&, Julie Dam5&, Vincent Prévot1&*
1 Univ. Lille, Inserm, CHU Lille, Laboratory of Development and Plasticity of the Neuroendocrine Brain, Lille Neuroscience & Cognition, UMR-S1172, EGID, DISTALZ, F-59000 Lille, France
2 CIMUS, Universidade de Santiago de Compostela-Instituto de Investigación Sanitaria, Santiago de Compostela, 15782, Spain- CIBER Fisiopatología de la Obesidad y Nutrición (CIBERobn), 15706, Spain
3 Univ. Lille, Inserm, CHU Lille, Institut Pasteur de Lille, CNRS, U1283 – UMR 8199 – EGID, F-59000 Lille, France
4 Centre National de la Recherche Scientifique, Université de Strasbourg, Institut des Neurosciences Cellulaires et Intégratives, F-67000 Strasbourg, France.
5 Institut Cochin, Inserm U1016, CNRS UMR 8104, University Paris Descartes, Sorbonne Paris Cité, Paris, France
6 Experimental Pharmacology, Center for Molecular Signaling (PZMS), Saarland University School of Medicine, 66421, Homburg, Germany
7 Laboratory of Tumor Inflammation and Angiogenesis, Center for Cancer Biology, VIB, Department of Oncology, KU Leuven, Leuven, B3000, Belgium
8 Cisbio Bioassays, Parc Technologique Marcel Boiteux, BP84175, F-30200 Codolet, France
9 VIB-UGent Center for Medical Biotechnology, Gent, Belgium.
10 Institute for Experimental and Clinical Pharmacology and Toxicology, University of Lübeck, Lübeck, Germany
11 Division of Endocrinology, Diabetes, and Metabolism, Beth Israel Deaconess Medical Center and Harvard Medical School, Boston, MA, USA.
12 Flow core Facility, BioImaging Center of Lille, campus HU, UMS2014-US41, F-59000 Lille, France
# These authors contributed equally
& These authors jointly supervised this work
Nature Metabolism, 2021
https://doi.org/10.1038/s42255-021-00432-5