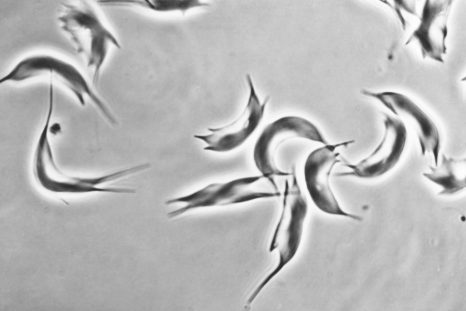
Sickle-shaped red blood cells (sickle cell disease) © Inserm/Chevance de Boisfleury, Anne-Marie
Teams from AP-HP, the University of Paris, Inserm, the Imagine Institute, the University of Paris-Est Créteil and the CEA conducted a clinical gene therapy study consisting of transplanting in the patient’s own genetically modified hematopoietic stem cells 1 . This phase I/II clinical trial, promoted by bluebird bio, was carried out in patients with sickle cell disease or transfusion-dependent beta thalassemia, common genetic diseases that affect red blood cells. The results of this work, coordinated by Pr Marina Cavazzana and Pr Philippe Leboulch, were published on January 24, 2022 in Nature Medicine .
As part of the HGB-205 phase I/II clinical trial, four b-thalassemia patients and three sickle cell patients aged 13 to 21 were treated with lentiviral gene therapy. They were followed for a median of 4.5 years after inclusion in the specific dedicated protocols LTF-303 (for b-thalassemia patients) and LFT-307 (for sickle cell patients).
According to the results, the patients with b-thalassemia all became “transfusion independent”, from the first month after the treatment, with a marked improvement in iron overload and a correction of the biological parameters linked to chronic anemia.
A remission of all the clinical symptoms 2 and a correction of the biological parameters sustained over time were obtained in two of the three sickle cell patients treated. A reduction in the transfusion rate was obtained for the third patient with sickle cell disease.
All of these results are maintained over time with more than 4.5 years of follow-up for three patients. No adverse effects linked to the use of the therapeutic lentiviral vector have been observed.
For patients with b-thalassemia, the reported long-term results show that gene therapy by gene addition has become a potentially usable curative option in all patients who do not have a compatible hematopoietic stem cell donor.
In the case of sickle cell disease, the correction of the biological parameters linked to the chronic anemia of two out of three patients provides proof in principle of its effectiveness and opens the way for the introduction of further improvements with the aim of obtaining the same result in all sickle cell patients treated.
Sickle cell disease and transfusion-dependent beta thalassemia
Sickle cell disease and transfusion-dependent b-thalassemia are common genetic diseases. They are therefore a major public health problem. These two chronic anemias are due to mutations in the gene encoding the beta (b) chain of adult hemoglobin (HbA).
Transfusion-dependent b-thalassemia is characterized by an absence (b 0 ) or a marked reduction (b + ) of the synthesis of b-globin chains, responsible for inefficient production of red blood cells and chronic hemolytic anemia and severe, requiring lifelong red blood cell transfusions. The resulting iron accumulation can cause heart failure, cirrhosis, liver cancer and multiple endocrine abnormalities.
Sickle cell disease results from the mutation of an amino acid in position 6 of the b-globin chain (E6V mutation), resulting in the polymerization of sickle cell hemoglobin HbS (hemoglobin “sickle” in English) once the molecules of oxygen (O 2 ) delivered. The polymerization of HbS is at the origin of painful vaso-occlusive crises characterized by a local obstruction of the blood circulation which can affect all the organs and of a chronic haemolytic anemia 3 . Repetition of vaso-occlusive crises and vascular damage affect several vital organs such as the lungs, kidneys, central nervous system and heart, with a significant reduction in the average lifespan of affected subjects.
As with almost all genetic diseases of the hematopoietic system, the only curative option is to transplant hematopoietic stem cells. This approach gives very good clinical results and low mortality when an HLA-compatible sibling donor is available. Unfortunately, a limited percentage of patients can benefit from this treatment (<20%). The use of partially matched donors greatly limits the chances of success and carries significant long-term morbidity, particularly in older patients.
Genetical therapy
Gene therapy, through the transplantation of stem cells from the patient himself, genetically modified, is a promising alternative. It carries low risks of immunological toxicity since no immunosuppressive treatment is required. It can be set up for each patient who needs it, the patient being his own donor.
Gene therapy by gene addition is the first strategy to have emerged in this indication by exploiting the ability of lentiviral vectors 4 to transfer complex genetic information into the genome of hematopoietic stem cells without the cells needing perform cell division.
The lentiviral vector used in this phase I/II clinical trial was developed by the team led by Prof. Philippe Leboulch. This vector allows the synthesis of a modified form of the b-globin chain (b T87Q ), a genetic modification which has a double interest: it gives it an anti-polymerizing property comparable to that of the gamma (g) chain of fetal hemoglobin (HbF) in sickle cell patients and allows its specific dosage in the blood of treated patients. Indeed, the b T87Q -globin chain can be distinguished from other globin chains by high pressure liquid chromatography, in particular from the b-globin chain derived from adult hemoglobins (HbA), produced endogenously in b- patients. thalassemia carriers of mutations b+ and present in transfused red blood cells.
1 Hematopoietic stem cells are nestled in the bone marrow and are at the origin of the different blood cells: red blood cells, white blood cells and platelets.
2 The set of pathological clinical signs that characterize a disease.
3 This is an often hereditary pathology that affects the red blood cells with, ultimately, a pathological reduction in their number incompatible with life and requiring regular blood transfusions, the frequency of which is dictated by the level of Hemoglobin and the clinical symptoms.
4 These are the shuttles of genetic information in the nucleus of cells derived from the HIV-1 virus from which the genetic elements that allow it to replicate and give rise to the infectious disease for which it is responsible have been removed. The elements which enable them to cross the nuclear membrane and to integrate in a stable manner into the genome of the target cells have, on the other hand, been preserved.
These contents could be interesting :
Medias
Sources
Long-Term Outcomes of Lentiviral Gene Therapy for the β-Hemoglobinopathies: the HGB-205 Trial
Nature Medicine
Elisa Magrin, Michaela Semeraro, Nicolas Hebert, Laure Joseph, Alessandra Magnani, Anne Chalumeau, Aurélie Gabrion, Cécile Roudaut, Jouda Marouene, Francois Lefrere, Jean-Sebastien Diana, Adeline Denis, Bénédicte Neven, Isabelle Funck-Brentano, Olivier Negre,Sylvain Renolleau, Valentine Brousse, Laurent Kiger, Fabien Touzot, Catherine Poirot, Philippe Bourget, Wassim El Nemer, Stéphane Blanche, Jean-Marc Tréluyer, Mohammed Asmal, Courtney Walls, Yves Beuzard, Manfred Schmidt, Salima Hacein-Bey-Abina, Vahid Asnafi, Isabelle Guichard, Maryline Poirée, Fabrice Monpoux, Philippe Touraine, Chantal Brouzes, Mariane de Montalembert, Emmanuel Payen, Emmanuelle Six, Jean-Antoine Ribeil, Annarita Miccio, Pablo Bartolucci, Philippe Leboulch & Marina Cavazzana