Contact Presse
AFM-Téléthon
Stéphanie Bardon, Géraldine Broudin, Gaëlle Monfort
Tél : 01 69 47 28 28 - rf.nohteneg.mfa@esserp Inserm rf.mresni@esserpUne publication dans le cadre des recherches sur la dystrophie musculaire facio-scapulo-humérale (FSHD), une des dystrophies musculaires les plus fréquentes, vient de paraître dans PLOS Genetics. L’équipe de Françoise Helmbacher de l’Institut de biologie du développement de Marseille (CNRS/ Aix-Marseille Université), soutenue par l’AFM-Téléthon en collaboration avec des chercheurs de l’Inserm, a identifié le gène FAT1 comme un nouvel acteur dans le développement des muscles et particulièrement chez les malades atteints de FSHD.
Le gène FAT1 permet la synthèse de la protocadhérine FAT1 qui est une protéine impliquée dans divers processus morphogénétiques. Ce gène agit dans les cellules musculaires en développement. Chez la souris, son ablation entraîne des défauts des muscles de la face et de la région des épaules, ainsi qu’une fonte musculaire précoce au stade adulte. En plus des anomalies musculaires, la dérégulation du gène FAT1 entraîne des défauts vasculaires dans la rétine et des malformations de l’oreille interne. L’ensemble de ces défauts ressemble fortement aux symptômes rencontrés dans la FSHD chez l’homme.
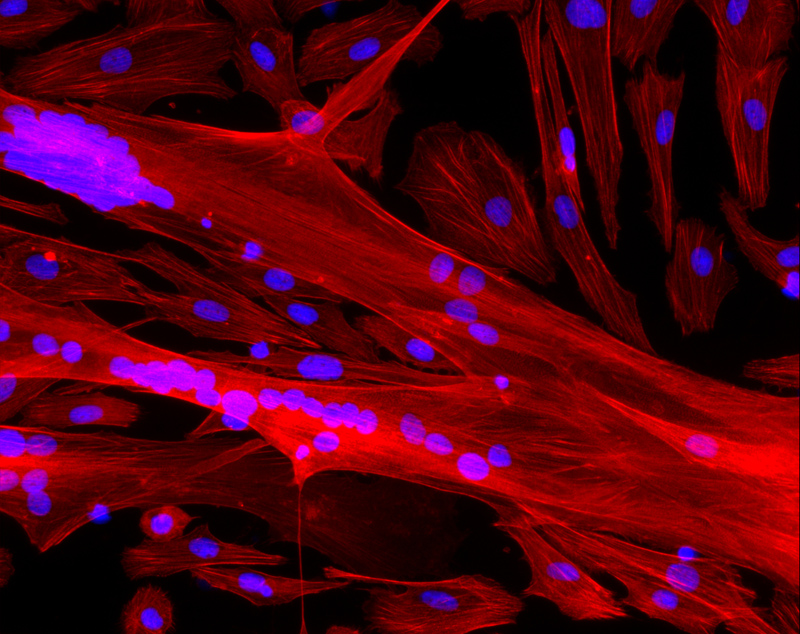
© Inserm/UPMC/CNRS/AIM
Les chercheurs ont donc exploré le lien entre la FSHD et ce gène FAT1 chez l’Homme. Ils ont retrouvé une baisse significative du niveau d’expression du gène dans les muscles FSHD au stade fœtal et des anomalies de ce gène sont retrouvées plus fréquemment chez les patients FSHD que chez les personnes non atteintes. Cette association concerne non seulement la forme classique (FSHD1), attribuée à une anomalie sur le chromosome 4 impliquant un fonctionnement excessif du gène DUX4, mais aussi les cas de FSHD non associés à DUX4. Les chercheurs ont montré que le gène DUX4 est également capable d’exercer un effet de répression du gène FAT1 dans des cellules musculaires.
Ainsi, sous l’apparente complexité de cette maladie, ces découvertes placent le gène FAT1 comme un dénominateur commun des différents mécanismes génétiques responsables de la FSHD, et en conséquence comme une nouvelle cible thérapeutique. L’ablation sélective du gène FAT1 chez la souris représente un moyen unique de mimer fidèlement les symptômes de la FSHD chez l’homme. Ce modèle animal va permettre de renforcer les connaissances sur la pathogénèse de cette maladie.
L’étude a été menée par l’équipe de Françoise Helmbacher de l’Institut de biologie du développement de Marseille (CNRS/ Aix-Marseille Université), en collaboration avec le laboratoire de Nicolas Levy (Inserm/ Aix-Marseille Université UMR 910, La Timone, Marseille, équipe Marc Bartoli), avec les participations de Julie Dumonceaux (Institut de Myologie, Paris), de Flavio Maina (IBDM), Frederique Magdinier (Inserm UMR 910), et Linda Geng (Fred Hutchinson Cancer Research Center, USA).
AFM-Téléthon
Stéphanie Bardon, Géraldine Broudin, Gaëlle Monfort
Tél : 01 69 47 28 28 - rf.nohteneg.mfa@esserp Inserm rf.mresni@esserp