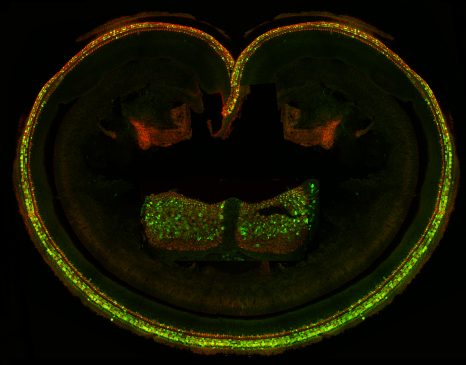
The cochlear sensory epithelium (organ of Corti), and the vestibular sensory epithelia, ampullar cristas (inset) of the inner ear ijntected with the AAV8 virus producing GFP and the SANS protein. The cochlear and vestibular sensory cells were immunostained for GFP and myosin VI and analyzed with a confocal microscope. The green and orange cells produce the protein of the therapeutic gene.
© Institut Pasteur
Scientists from the Institut Pasteur, Inserm, the CNRS, Collège de France, University Pierre et Marie Curie, and University Clermont Auvergne*, have recently restored hearing and balance in a mouse model of Usher syndrome type 1G (USH1G) characterized by profound congenital deafness and vestibular disorders caused by severe dysmorphogenesis of the mechanoelectrical transduction apparatus of the inner ear’s sensory cells. By locally injecting the USH1G gene, critical for the formation and maintenance of the hair bundle, the mechanosensory antenna of these cells, the scientists successfully restored the function and the structure of this apparatus, resulting in the first recovery of hearing and balance by gene therapy for this mouse model. These findings, published in the journal PNAS, open up new possibilities for the development of gene therapy treatments for hereditary forms of deafness.
Hearing loss, sometimes associated with other disorders such as balance defects, is the most common sensory deficit, affecting more than 280 million people worldwide, according to WHO. In France, one child in 700 is born with severe or profound hearing loss, and one in every 1,000 will lose their sense of hearing before adulthood.
Over the past 20 years, scientists have made remarkable progress in deciphering the genetic origins of congenital hereditary hearing loss, which is usually caused by inner ear dysfunction. The inner ear comprises the hearing organ or cochlea, together with the five balance organs (the saccule, utricle and three semicircular canals), which contain the sensory cells, or hair cells, that detect mechanical vibrations and convert them into electrical signals. To date, mutations in more than 100 genes have been associated with inner ear defects, and it is estimated that mutations in more than 100 genes can cause genetic forms of deafness.
The various hereditary forms of hearing loss include Usher syndrome type 1 (USH1), a particularly severe clinical form of deaf-blindness, and specifically the USH1G genetic form. USH1G patients are profoundly deaf and have no balance function at birth, and they subsequently suffer from prepubertal-onset sight loss leading to blindness. USH1G syndrome is due to mutations in the gene encoding the scaffold protein sans, which is essential for the cohesion of the hair bundle of the inner ear hair cells.
Patients with hearing loss and balance dysfunction are currently fitted with auditory prostheses and may be given balance rehabilitation therapy, but the outcomes are variable. One possible alternative for treating such hereditary inner ear defects is gene therapy. This approach entails transferring a healthy (non-mutant) copy of the defective gene to restore the expression of the missing protein. So far, gene therapy attempts have only resulted in partial improvements of hearing in mouse models of specific human deafness forms that did not include severe anomalies in hair cell structure.
In this context, scientists from the Institut Pasteur, Inserm, the CNRS, Collège de France, University Pierre et Marie Curie, and University Clermont Auvergne*, have now succeeded in restoring hearing and balance in a mouse model of USH1G syndrome using gene therapy. With a single local injection of the USH1G gene just after birth, the scientists observed a restoration of the structure and mechanosensory function of the inner ear hair bundles – profoundly damaged before birth –, resulting in a long-term partial recovery of hearing, and complete recovery of vestibular function in these mice. These results unexpectedly establish that inner ear defects due to major morphogenetic abnormalities of the hair bundle can be reversed even after birth, with durable efficacy, by gene therapy.
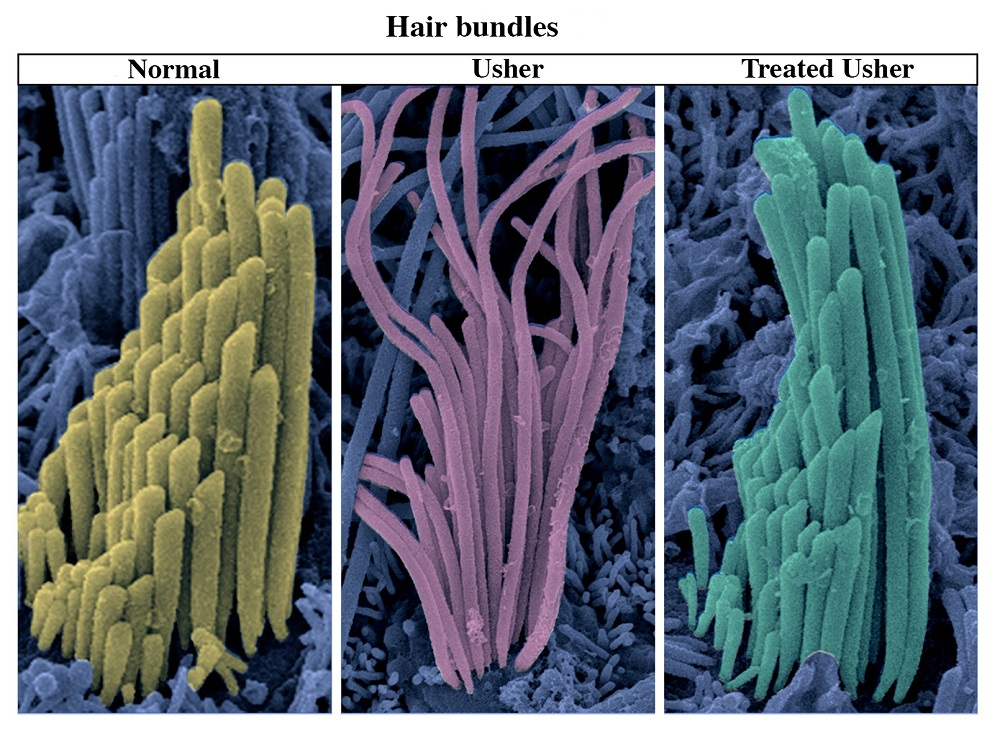
Hair bundles of vestibular sensory cells analyzed using scanning electron microscopy. The image shows a normal hair bundle with its characteristic “staircase” pattern (in yellow), a defective Usher type 1G hair bundle (in pink) and a treated Usher type 1G hair bundle (in green), whose normal/characteristic form was restored with gene therapy.
© Institut Pasteur
The scientists injected the USH1G gene into the inner ear using the innocuous AAV8 virus, which enabled them to specifically target the hair cells. The expression of the therapeutic gene was detected 48 hours after injection. The team demonstrated that a single injection to restore the production and localization of the missing protein in hair cells successfully improved hearing and balance functions in the young mice. These findings suggest that the therapeutic protein was able to interact normally with its binding partners among the USH1 molecular complex (the proteins cadherin 23, protocadherin 15, myosin VIIA and harmonin), as required for the mechanoelectrical transduction apparatus of the hair bundle to function correctly.
As Saaïd Safieddine, CNRS Director of Research at the Institut Pasteur and co-senior author of the study with Prof. Christine Petit, explains, “we have just shown that it is possible to partially correct a specific form of hereditary hearing loss accompanied by balance problems using local gene therapy performed after the embryogenesis of the ear, which is primarily affected by the mutation responsible for the disorder. This suggests that the time window for effectively treating USH1 syndrome using gene therapy may be larger than initially thought.”
This study represents a significant step towards the development of clinical trials in gene therapy for the curative treatment of hereditary deafness and balance loss in humans.
*From the Genetics & Physiology of Hearing Laboratory (Institut Pasteur/Inserm/UPMC), the Genes, Synapses and Cognition Laboratory (CNRS/Institut Pasteur, the Center for Neurophysics, Physiology and Pathology (CNRS/Paris-Descartes University), and the Sensory Biophysics Laboratory (University Clermont Auvergne).
These contents could be interesting :
Medias
Sources
Local gene therapy durably restores vestibular function in a mouse model of Usher syndrome type 1G, PNAS, 5 septembre 2017 Alice Emptoz (a,b,c), Vincent Michela (b,c), Andrea Lellia (b,c), Omar Akil (d), Jacques Boutet de Monvela (b,c), Ghizlene Lahloua (b,c), Anaïs Meyera (b), Typhaine Duponta (b,c), Sylvie Nouaillea (b,c), Elody Ey €, Filipa Franca de Barros (f), Mathieu Beraneck (f), Didier Dulon (g), Jean-Pierre Hardelina (b,c), Lawrence Lustig (h), Paul Avan (i), Christine Petita (b,c,j), and Saaid Safieddine (a,b,c).
(a) INSERM, UMR 1120, Paris, France.
(b) Génétique et Physiologie de l’Audition, Institut Pasteur, 75015 Paris, France.
(c) Complexité du Vivant, Sorbonne Universités, Université Pierre-et-Marie-Curie, Université Paris VI, 75015 Paris, France.
(d) Otolaryngology-Head & Neck Surgery, University of California, San Francisco, CA 94117.
(e) Unité de Génétique Humaine et Fonctions Cognitives, Institut Pasteur, CNRS UMR 3571, 75015 Paris, France.
(f) Centre de Neurophysique, Physiologie, et Pathologie, CNRS UMR 8119, Université Paris-Descartes, 75006 Paris, France.
(g) Laboratoire de Neurophysiologie de la Synapse Auditive, Bordeaux Neurocampus, INSERM, UMR 1120, Université de Bordeaux, 33076 Bordeaux, France.
(h) Columbia University School of Medicine and New York Presbyterian Hospital, New York, NY 10034.
(i) Laboratoire de Biophysique Sensorielle, Faculté de Médecine, Université d’Auvergne, Biophysique Médicale, Centre Jean Perrin, 63000 Clermont-Ferrand, France.
(j) Collège de France, 75005 Paris, FranceDOI : 10.1073/pnas.1708894114