

Contact Presse
Service de presse de l'Institut Pasteur
Myriam Rebeyrotte
01 45 68 81 01
Aurélie Perthuison
01 45 68 89 28
Service de presse de l'Inserm
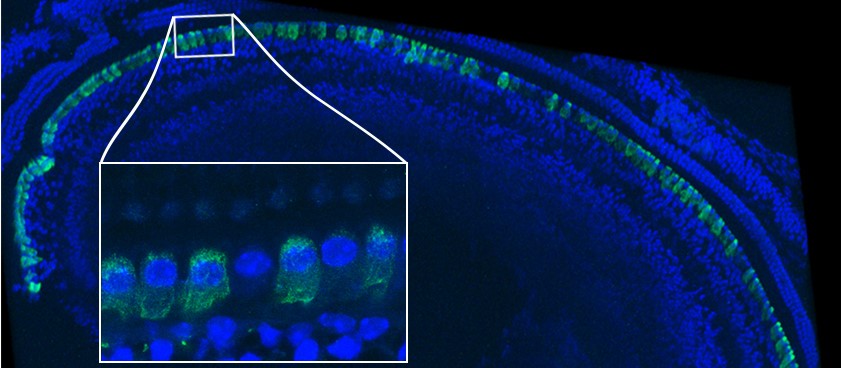
Image par immunofluorescence de l’épithélium sensoriel d’une cochlée de souris traitée par thérapie génique © Institut Pasteur
Des chercheurs de l’Institut Pasteur, de l’Inserm, du CNRS, du Collège de France, de Sorbonne Université et de l’Université Clermont Auvergne, et en collaboration avec les universités de Miami, de Columbia et de San Francisco, viennent de parvenir à restaurer l’audition au stade adulte chez un modèle murin de la surdité DFNB9, un trouble auditif représentant l’un des cas les plus fréquents de surdité congénitale d’origine génétique. Les sujets atteints de surdité DFNB9 sont sourds profonds, étant dépourvus du gène codant pour l’otoferline, protéine essentielle à la transmission de l’information sonore au niveau des synapses des cellules sensorielles auditives. Grâce à l’injection intracochléaire de ce gène chez un modèle murin de cette surdité, les chercheurs sont parvenus à rétablir la fonction de la synapse auditive et les seuils auditifs des souris à un niveau quasi-normal. Ces résultats, publiés dans la revue PNAS, ouvrent la voie à de futurs essais de thérapie génique chez des patients atteints de DFNB9.
Plus de la moitié des cas de surdité congénitale profonde non syndromique ont une cause génétique, et la plupart (~ 80%) de ces cas sont dus à des formes autosomiques récessives de surdité (DFNB). Les implants cochléaires sont actuellement la seule option permettant une récupération auditive chez ces patients.
Les virus adéno-associés (AAV) sont parmi les vecteurs les plus prometteurs pour le transfert de gènes dans le but de traiter des maladies humaines. La thérapie génique basée sur les AAV est une option thérapeutique prometteuse pour le traitement des surdités, mais son application est limitée par une fenêtre thérapeutique potentiellement courte. En effet, chez l’humain, le développement de l’oreille interne s’achève in utero et l’audition débute à environ 20 semaines de gestation. En outre, les formes génétiques de surdité congénitale sont généralement diagnostiquées au cours de la période néonatale. Les approches de thérapie génique dans les modèles animaux doivent donc en tenir compte et l’efficacité du gène thérapeutique doit être démontrée pour une injection du gène effectuée après la mise en place de l’audition. La thérapie doit alors conduire à la réversion de la surdité déjà installée. Dans ce but, l’équipe dirigée par Saaïd Safieddine, chercheur CNRS au sein de l’unité de Génétique et de physiologie de l’audition (Institut Pasteur/ Inserm), et coordinateur du projet, a utilisé un modèle murin de DFNB9, une forme de surdité humaine représentant 2 à 8 % de l’ensemble des cas de surdité génétique congénitale.
La surdité DFNB9 est due à des mutations dans le gène qui code pour l’otoferline, une protéine jouant un rôle majeur dans la transmission de l’information sonore au niveau des synapses des cellules ciliées internes[1]. Les souris mutantes dépourvues d’otoferline sont sourdes profondes en raison d’une défaillance complète de la libération de neurotransmetteur par ces synapses en réponse à la stimulation sonore, et ce malgré l’absence d’anomalie décelable de l’épithélium sensoriel. Les souris DFNB9 constituent donc un modèle approprié pour tester l’efficacité de la thérapie génique virale lorsqu’elle est administrée à un stade mature. Cependant, la capacité limitée d’empaquetage de l’ADN par les AAV (environ 4,7 kilo-bases (kb)), rend difficile l’utilisation de cette technique pour des gènes dont la séquence codante (ADNc) dépasse 5 kb, tel que le gène Otof codant pour l’otoferline, dont la séquence codante est de 6 kb. Les chercheurs ont surmonté cette limitation en adaptant une approche d’AAV, dite duale, parce qu’elle utilise deux vecteurs recombinants différents, l’un contenant la partie 5’ et l’autre la partie 3’ de l’ADNc de l’otoferline.
Une seule injection de la paire de vecteurs dans la cochlée de souris mutantes à des stades adultes a permis de reconstituer la séquence codante de l’otoferline par recombinaison des segments d’ADN 5′ et 3′, conduisant à la restauration durable de l’expression de l’otoferline dans les cellules ciliées internes, puis à une restauration de l’audition.
Les résultats obtenus par les chercheurs suggèrent que la fenêtre thérapeutique pour le transfert de gène local chez les patients atteints de surdité congénitale DFNB9 pourrait être plus large que prévu, et donnent l’espoir de pouvoir étendre ces résultats à d’autres formes de surdité. Ces résultats font l’objet d’une demande de brevet.
En plus des institutions citées dans le premier paragraphe, ce travail a été financé par la Fondation pour la recherche médicale, l’Union européenne (TREAT RUSH) et par l’Agence nationale de la recherche (EargenCure et LabEx Lifesenses).
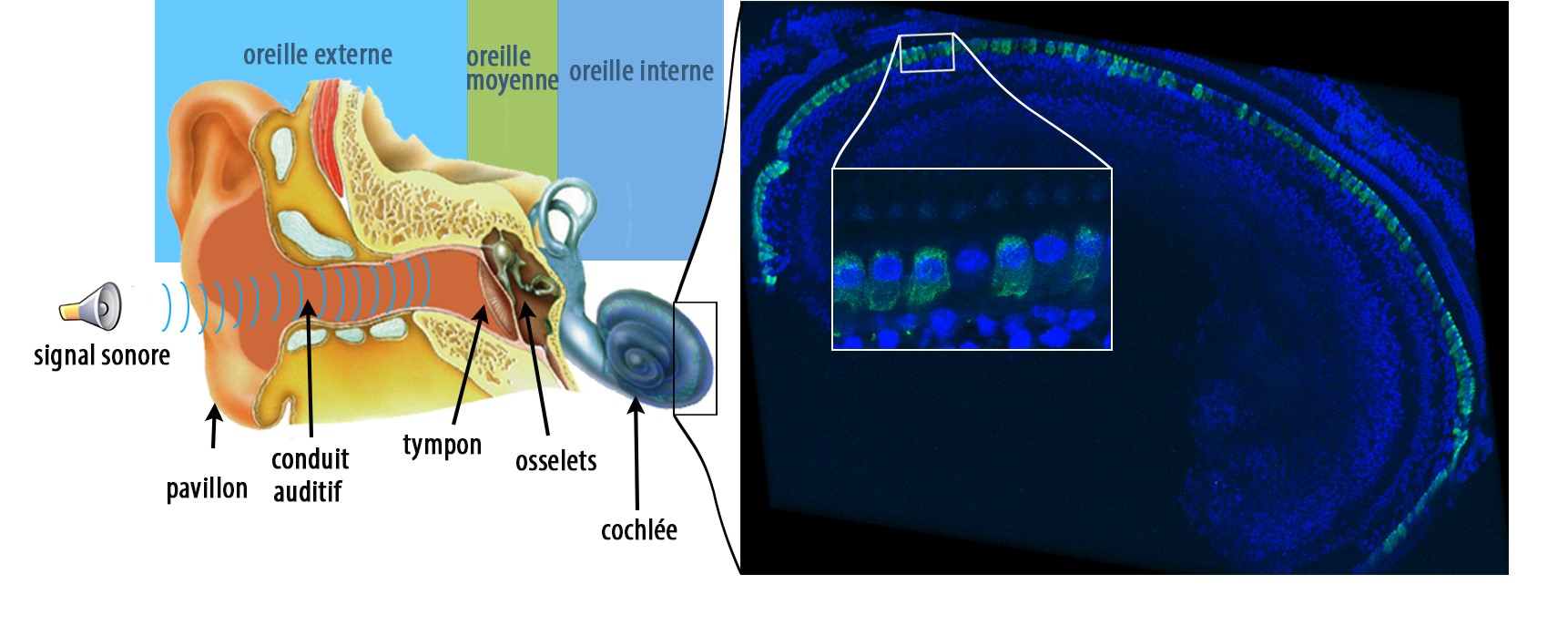
La figure de gauche est une représentation schématique de l’oreille humaine : les vibrations sonores sont collectées par l’oreille externe composée du pavillon et du conduit auditif. L’oreille moyenne, composée du tympan et des osselets, transmet les vibrations sonores à l’oreille interne où se trouve la cochlée, organe de l’audition responsable de la transmission du message auditif au système nerveux central. La figure de droite montre une image par immunofluorescence de l’épithélium sensoriel d’une cochlée de souris traitée, où les cellules ciliées internes ont été marquées en vert pour révéler l’otoferline. L’otoferline est détectée dans la quasi-totalité de ces cellules. La zone à fort grossissement dans l’encadré montre une cellule ciliée interne qui n’a pas été transduite. © Institut Pasteur
Service de presse de l'Institut Pasteur
Myriam Rebeyrotte
01 45 68 81 01
Aurélie Perthuison
01 45 68 89 28
Service de presse de l'Inserm
Dual AAV-mediated gene therapy restores hearing in a DFNB9 mouse model, PNAS, xx janvier 2019
Omar Akil*§§, Frank Dyka†, Charlotte Calvet‡,§,¶, Alice Emptoz‡,§,¶, Ghizlene Lahlou‡,§,¶, Sylvie Nouaille‡,§,¶, Jacques Boutet de Monvel‡,§,¶, Jean-Pierre Hardelin‡,§,¶, William Hauswirth†, Paul Avan#, Christine Petit‡,§,¶,**,¶¶, Saaid Safieddine‡,§,¶,††,¶¶, Lawrence R. Lustig‡‡,§§
*Department of Otolaryngology-Head & Neck Surgery, University of California San Francisco, San Francisco, CA, USA
†Department of Ophthalmology, University of Florida, College of Medicine, Gainesville, FL, USA
‡Genetics and Physiology of Hearing Laboratory, Institut Pasteur, Paris, France.
¶Sorbonne Universités, UPMC Université Paris 06, Complexité du Vivant, Paris, France
#Laboratoire de Biophysique Sensorielle, Faculté de Médecine, Université Clermont Auvergne, Biophysique Médicale, Centre Jean Perrin, Clermont-Ferrand, France.
**Collège de France, Paris, France
††Centre National de la Recherche Scientifique, France
‡‡Department of Otolaryngology-Head & Neck Surgery, Columbia University Medical Center, and New York Presbyterian Hospital, USA
¶¶ Corresponding authors