Une équipe internationale, incluant en France, des chercheurs de l’Inserm, du CNRS et de l’Université de Strasbourg réunis au sein de l’IGBMC[1] lève le voile sur les mécanismes moléculaires à l’origine des troubles cardiaques de la dystrophie myotonique, une maladie génétique touchant un individu sur 8 000. Cette nouvelle étude publiée cette semaine dans Nature Communications pourrait contribuer à la découverte d’un traitement.
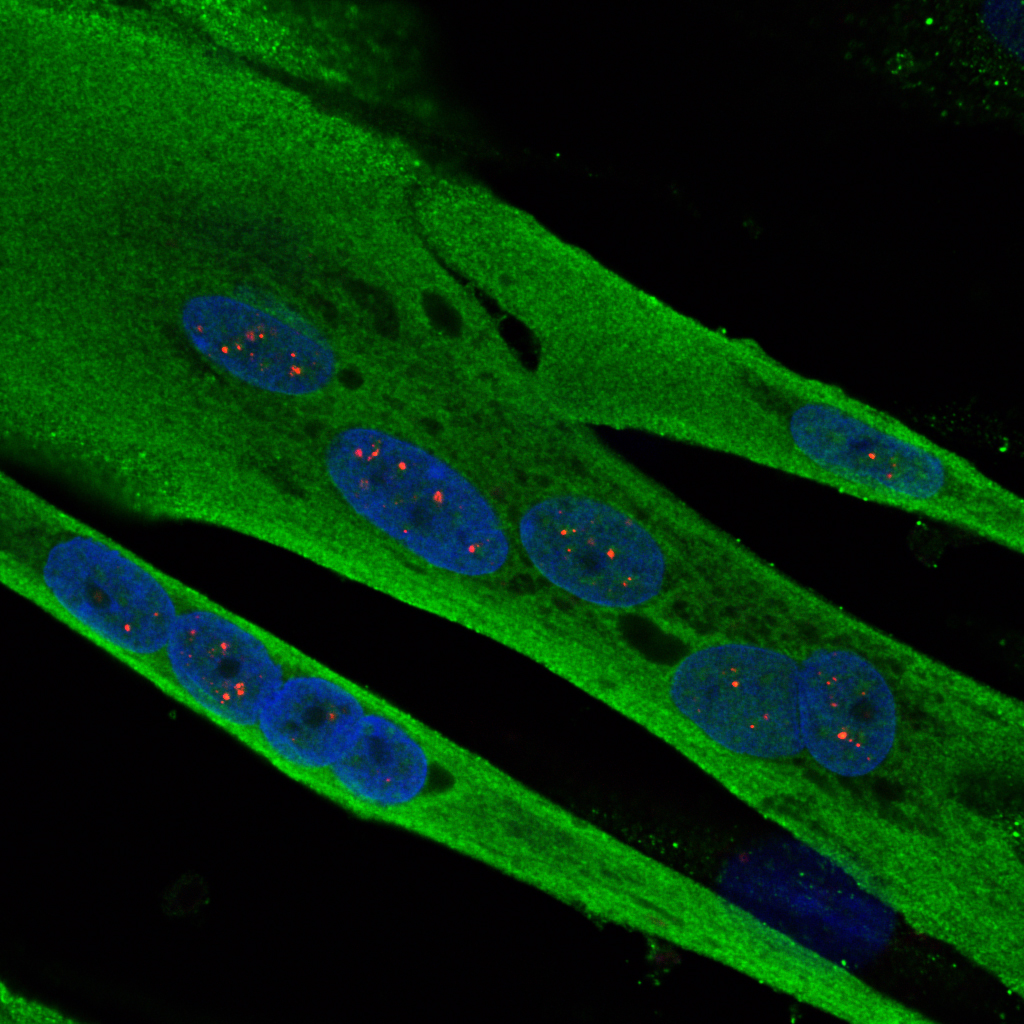
Cellules musculaire de patient atteint de dystrophie myotonique (ADN nucleaire en bleu, aggregats d’ARN typique de la dystrophie myotonique en rouge et cytoplasme en vert)
(c) Inserm/IGBMC
La dystrophie myotonique, aussi connue sous le nom de maladie de Steinert, est la forme adulte la plus commune de dystrophie musculaire. Les patients atteints de cette affection génétique souffrent d’un affaiblissement des muscles squelettiques mais aussi d’arythmie et d’autres troubles cardiaques. Il s’agit d’une maladie particulièrement invalidante pour laquelle il n’existe pour le moment aucun traitement.
La dystrophie myotonique est due à une mutation conduisant à l’expression d’ARN contenant de longues répétitions du tri-nucléotide CUG. Ces ARN mutés s’accumulent et altèrent la régulation de l’épissage alternatif[2] de nombreux gènes. Malgré l’importance des travaux déjà effectués sur cette maladie, de nombreux points restent à élucider. C’est le cas de l’origine des arythmies et autres troubles cardiaques, qui représentent la deuxième cause de décès dans cette maladie.
Dans cette nouvelle étude, les chercheurs ont identifié de nouvelles altérations d’épissage dans les ARN messagers des échantillons de cœur de patients atteints. Parmi ces nombreuses altérations, les biologistes ont établi que celles concernant le canal sodique cardiaque (SCN5A) étaient fondamentales pour comprendre les troubles cardiaques de ces patients.
Les scientifiques ont alors éclairci les mécanismes moléculaires conduisant à l’altération de SCN5A chez ces patients. Une collaboration avec l’équipe de Denis Furling l’Institut de myologie à Paris, a permis de reproduire ces altérations cardiaques dans un modèle de souris.
« La prochaine étape serait de voir si en rétablissant un épissage correct de SCN5A, nous réussissons à retrouver aussi un fonctionnement normal du cœur », explique Nicolas Charlet-Berguerand, directeur de recherche Inserm, qui a coordonné ce travail. Les chercheurs espèrent que cette avancée donnera un nouvel élan à la recherche sur cette maladie rare.
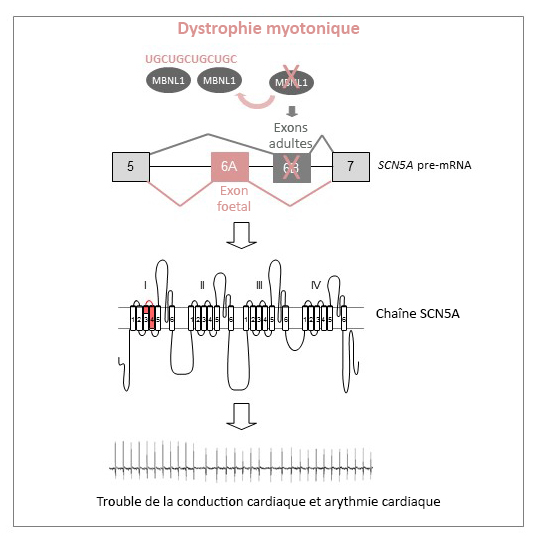
Modèle d’épissage alternatif du canal sodique cardiaque (SCN5A) dans la dystrophie myotonique.
(c) Inserm/IGBMC
Ce travail a été financé par l’association française contre les myopathies (AFM), l’European research council (ERC), le programme européen E-rare (ANR), l’Inserm et le Labex-INRT (ANR)
[1] Institut de génétique et de biologie moléculaire et cellulaire (Inserm/CNRS/Université de Strasbourg)
[2] Chez les eucaryotes, il s’agit d’un processus par lequel l’ARN transcrit à partir d’un gène peut subir différentes étapes de coupure et ligature conduisant à l’élimination de diverses régions. Ce procédé permet la production à partir d’un même gène de protéines ayant des propriétés distinctes.
Ces contenus pourraient aussi vous intéresser :

Contacts
Sources
Splicing misregulation of SCN5A contributes to cardiac-conduction delay and heart arrhythmia in myotonic dystrophyFernande Freyermuth1,*,w,Frederique Rau2,*, Yosuke Kokunai3, Thomas Linke4, Chantal Sellier1, Masayuki Nakamori3,Yoshihiro Kino5, Ludovic Arandel2, Arnaud Jollet2, Christelle Thibault1, Muriel Philipps1, Serge Vicaire1, Bernard Jost1,Bjarne Udd6,7,8, John W. Day9, Denis Duboc10, Karim Wahbi10, Tsuyoshi Matsumura11, Harutoshi Fujimura11,Hideki Mochizuki3, François Deryckere12, Takashi Kimura13, Nobuyuki Nukina14, Shoichi Ishiura15, Vincent Lacroix16,Amandine Campan Fournier17, Vincent Navratil18, Emilie Chautard19, Didier Auboeuf19, Minoru Horie20, Keiji Imoto21,Kuang-Yung Lee22, Maurice S. Swanson23, Adolfo Lopez de Munain24, Shin Inada25, Hideki Itoh20, Kazuo Nakazawa25,Takashi Ashihara20, Eric Wang23, Thomas Zimmer4, Denis Furling2, Masanori P. Takahashi3 & Nicolas Charlet-Berguerand11 Department of Translational medicine and neurogenetics, IGBMC, CNRS UMR7104, INSERM U964, Université de Strasbourg, Illkirch 67400, France.
2Sorbonne Universités UPMC Univ Paris 06, Inserm, CNRS, Centre de Recherche en Myologie UMRS974/FRE3617, Institut de Myologie, GH Pitié-Salpêtrière, Paris 75013, France.
3Department of Neurology, Osaka University Graduate School of Medicine, Osaka 565-0871, Japan.
4nDepartment of Physiology, Friedrich Schiller U niversity Hospital, Jena 07743, Germany.
5 Department of Bioinformatics and Molecular Neuropathology, Meiji Pharmaceutical University, Kiyose 205-8588, Japan.
6 Neuromuscular Research Center, Tampere University and University Hospital, Tampere 33520, Finland.
7 Department of Medical Genetics, Folkhalsan Institute of Genetics, Helsinki University, Helsinki 00250, Finland.
8 Department of Neurology, Vaasa Central Hospital, Vaasa 65130, Finland.
9 Department of Neurology, Stanford University, Stanford, California 94304, USA.
10 Service de Cardiologie, Université Paris-Descartes, Hôpital Cochin, AP-HP, Paris 75014, France.
11 Department of Neurology, Toneyama National Hospital, Toyonaka 560-8552, Japan.
12 CNRS UMR7175, Ecole Supérieure de Biotechnologies de Strasbourg, Illkirch 67400, France.
13 Division of Neurology, Hyogo Medical College, Nishinomiya 663-8501, Japan.
14 Laboratory of Structural Neuropathology, Doshisha University Graduate School of Brain Science, Kyoto 610-0394, Japan.
15Graduate School of Arts and Sciences, University of Tokyo , To k y o 1 5 3 - 8 9 0 2 , J a p a n .
16 Université Lyon 1, CNRS, UMR5558 LBBE, Villeurbanne 69622, France.
17Hospices civils de Lyon, Laboratoire de cytogénétique constitutionnelle, Bron 69500, France.
18Pole Rhône Alpes de Bio-informatique, Université Lyon 1, Bâtiment Gregor Mendel, Villeurbanne 69100, France.
19Centre de Recherche en Cancérologie deLyon,Lyon69373,France.
20 Department of Cardiovascular and Respiratory Medicine, Shiga Medical University, Otsu 520-2192, Japan.
21 Department of Information Physiology, National Institute for Physiological Sciences, Okazaki 444-8585, Japan.
22 Department of Neurology, Chang Gung Memorial Hospital, Keelung 20401, Taiwan.
23 Department of Molecular Genetics and Microbiology, Center for NeuroGenetics and the Genetics Institute, University of Florida, College of Medicine, Gainesville, Florida 32610, USA.
24 Department of Neurology, Hospital Universitario DONOSTIA, Neuroscience Area, Institute Biodonostia CIBERNED and University of Basque CountryUPV-EHU, San Sebastian20014, Spain.
25 Laboratory of Biomedical Sciences and Information Management, National Cerebral and Cardiovascular Center Research Institute, Osaka 565-8565, Japan. * These authors contributed equally to the workNature Communications, avril 2016