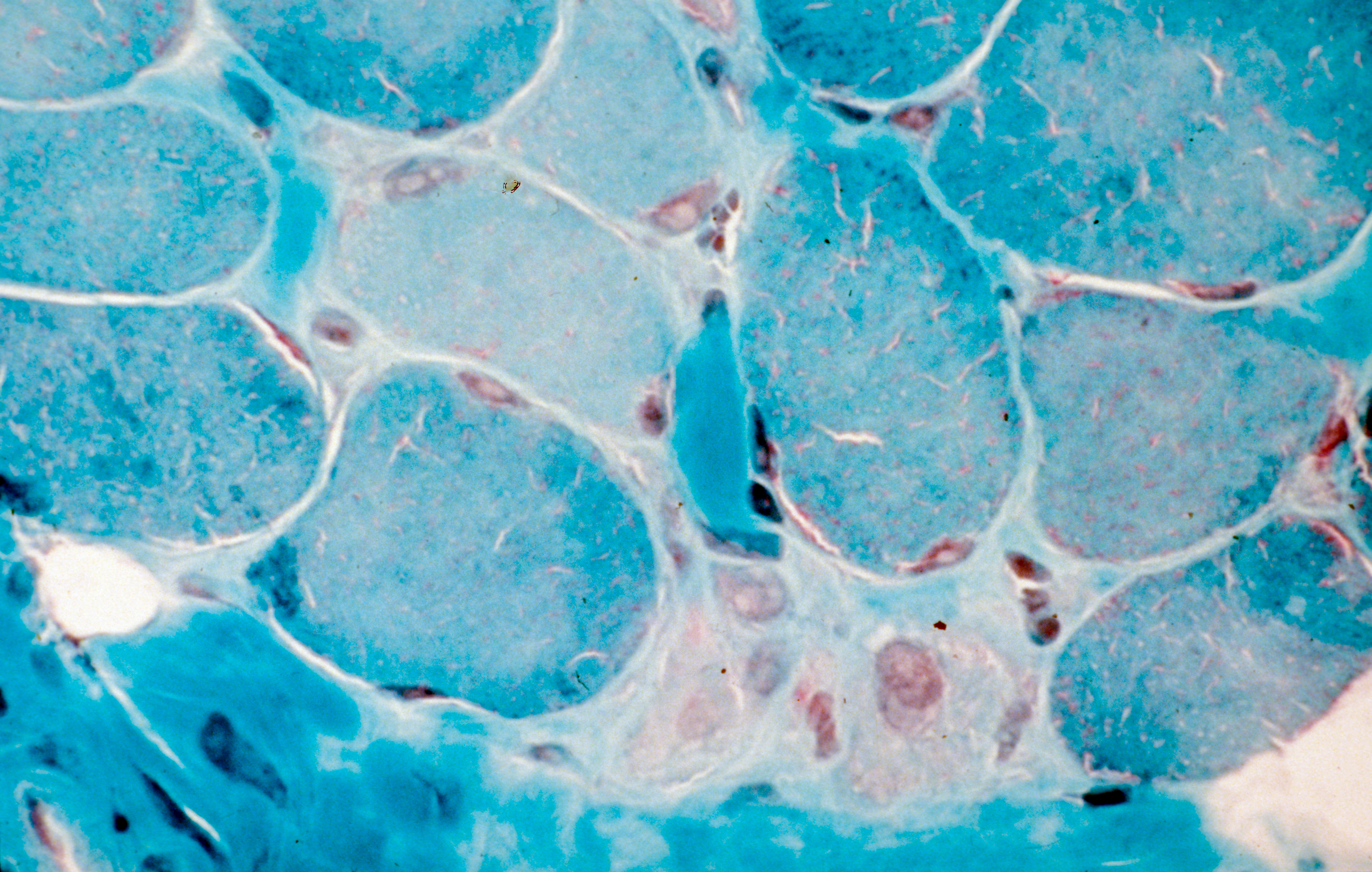
Coupe transversale de muscle humain, régénération de fibres musculaires après un traitement de la myopathie de Duchenne. Crédits: Inserm/Fardeau, Michel
L’équipe « Myopathies inflammatoires et thérapies innovantes ciblées » de l’Institut de Myologie, dirigée par le Pr Olivier Benveniste, a mis en évidence une nouvelle classification des myosites, maladies inflammatoires du muscle. Désormais, 4 nouveaux types de myosites, prenant en compte tous les critères cliniques des patients, sont définis. Ces travaux impliquant des équipes de recherche de l’Institut de Myologie, de l’Inserm, de l’AP-HP et de Sorbonne Université, publiés ce jour dans la revue JAMA, ouvrent la voie à un diagnostic fiable et à des traitements personnalisés.
Les myosites (ou myopathies inflammatoires) constituent un groupe de maladies rares auto-immunes du muscle, c’est-à-dire des maladies dans lesquelles le système immunitaire, chargé de protéger l’organisme contre des attaques extérieures (microbe, virus…), se dérègle et s’attaque à l’organisme (ici le muscle). Elles concernent entre 3 000 et 5 000 adultes et enfants en France.
Si toutes les myosites ont une composante auto-immune, chacune possède des mécanismes de déclenchement qui lui sont propres. Jusqu’à présent, la classification identifiait 3 types de myosites (polymyosite, dermatomyosite, myosite à inclusions) selon un système de classification établi en 1975 puis mis à jour en 2017 (critères ACR,/EULAR des Rhumatologues) et fondé essentiellement sur des critères cliniques et histologiques. Le Pr Olivier Benveniste, responsable de l’équipe « Myopathies inflammatoires et thérapies innovantes ciblées » à l’Institut de Myologie, suivant quotidiennement des patients depuis 20 années à l’hôpital Pitié-Salpêtrière, AP-HP, a identifié des erreurs diagnostiques graves liées à cette classification incomplète et, de fait, non-homogène, de ces pathologies générant parfois même des erreurs dans le traitement donné aux patients. Certains patients diagnostiqués par erreur comme ayant une myosite à inclusions ont pu être ainsi traités avec une forte dose de corticoïdes alors que ces derniers aggravent leur état.
C’est pourquoi, avec son équipe et en collaboration avec le centre de référence des maladies neuromusculaires de l’Institut de Myologie, il a lancé une étude sur 260 patients dont il a recueilli et analysé toutes les caractéristiques cliniques et notamment la présence d’auto-anticorps, parfois causes ou conséquences de la maladie. Par des méthodes statistiques innovantes et sans a priori, c’est à dire l’algorithme mathématique agrège les patients qui se ressemblent (analyse en cluster) sans intervention des chercheurs, ces derniers ont mis en évidence une nouvelle classification avec 4 grands types de myosites : myosite à inclusions, dermatomyosite, myopathie nécrosante auto-immune, syndrome des anti-synthétases (les polymyosites ne constituant plus un type de myosite en tant que tel).
Caractéristiques des 4 types :
▪ Myosite à inclusions : Cette myosite affecte plus souvent les hommes de plus de 60 ans. Elle est lentement progressive mais induit finalement un déficit moteur très handicapant. Elle touche plus particulièrement les quadriceps (muscle des cuisses qui servent à monter les escaliers, se relever d’une chaise, être stable à la marche…), les muscles qui servent à fermer et serrer les mains et les muscles de la déglutition. Cette maladie résiste aux traitements immunosuppresseurs classiques comme les corticoïdes. Elle est due à la présence dans le muscle d’une réaction inflammatoire (la myosite) et d’un processus neurodégénératif apparenté à la maladie d’Alzheimer (donnant les inclusions).
▪ Dermatomyosite : Elle touche plus souvent les femmes. Les enfants peuvent être atteints. Un risque de cancer associé apparait chez les sujets les plus âgés (généralement après 60 ans). Outre la myosite avec qui entraine une faiblesse musculaire prédominante aux épaules, cette maladie est caractérisée par la présence de lésions dermatologiques typiques. Cette maladie est due à un dérèglement du système immunitaire mettant en jeu l’interféron de type 1 qui permet de se défendre contre les virus. De nouveaux traitements ciblant spécifiquement cette voie de l’interféron sont en cours de développement. Les anticorps spécifiques des dermatomyosites sont les anti-Mi2, anti-SAE, anti-NXP2, ou anti-TIF1gamma.
▪ Myopathie nécrosante auto-immune : Il s’agit d’une atteinte purement musculaire touchant les patients de tout âge. C’est la myosite qui en l’absence de traitement conduit à l’atrophie musculaire la plus sévère et handicapante. Cette maladie est liée à la présence de deux anticorps spécifiques anti-SRP ou anti-HMGCR qui attaquent et détruisent directement les muscles. Les anti-HMGCR peuvent apparaitre après la prise de statines. Le traitement vise ici à faire disparaitre ces anticorps.
▪ Syndrome des anti-synthétases : Cette maladie touche le muscle mais aussi les articulations (donnant un rhumatisme), et les poumons (donnant un essoufflement parfois sévère). Ici aussi, certains anticorps semblent responsables. Il s’agit des anti-Jo1, anti-PL7 ou anti-PL12.
Cette nouvelle classification est déterminante pour poser un diagnostic et proposer un traitement personnalisé aux malades.
«Nous nous sommes rendu compte que la classification actuelle des myosites n’était pas adaptée et pouvait souvent conduire à l’échec d’un traitement potentiel en raison de groupes de patients non-homogènes dans un même essai. Notre but était donc de définir une classification fondée des critères phénotypiques, biologiques et immunologiques afin de pouvoir mieux diagnostiquer les différents types de myosites et de trouver, à terme, des traitements adaptés pour les malades. Cette nouvelle classification devient une référence puisque même la FDA, qui utilisait jusqu’alors la classification américaine, recommande de se baser sur nos travaux. » explique le Pr Benveniste.
Ces contenus pourraient aussi vous intéresser :

Contacts
Sources
A New Classification System for Idiopathic Inflammatory Myopathies Based on Clinical Manifestations and Myositis-Specific Autoantibodies - Kubéraka Mariampillai, Benjamin Granger, Damien Amelin, Marguerite Guiguet, Eric Hachulla, François Maurier, Alain Meyer, Aline Tohmé, Jean-Luc Charuel, Lucile Musset, Yves Allenbach and Olivier Benveniste.Centre de Recherche en Myologie, Unité Mixte de Recherche Scientifique 974, Université Pierre et Marie Curie, Institut National de la Santé et de la Recherche Médicale, Paris, France (Mariampillai, Amelin, Allenbach, Benveniste); Département de Médecine Interne et Immunologie Clinique, Centre de Référence Maladies Neuro-Musculaires, Assistance Publique– Hôpitaux de Paris, Groupe Hospitalier Pitié-Salpêtrière, DHUi2B, Paris, France (Mariampillai, Allenbach, Benveniste); Département de Biostatistiques, Santé Publique et Information Médicale, Assistance Publique–Hôpitaux de Paris, Groupe Hospitalier Pitié-Salpêtrière, Paris, France (Granger); Institut Pierre Louis d'Epidémiologie et de Santé Publique, Université Pierre et Marie Curie– Paris 6, Sorbonne Universités, Groupe de Recherche Clinique–08, Epidémiologie et Evaluation des Maladies Ostéoarticulaires Inflammatoires et Systémiques, Paris, France (Granger); Sorbonne Universités, Institut Pierre Louis d’Epidémiologie et de Santé Publique, Université Pierre et Marie Curie–Paris 6, Unité Mixte de Recherche Scientifique 1136, Paris, France (Guiguet); Service de Médecine Interne, Centre Hospitalier Universitaire, Lille, France (Hachulla); Service de Médecine Interne, Hôpital Belle-Isle, Metz, France (Maurier); Département de Physiologie, Centre Hospitalier Universitaire de Strasbourg, Strasbourg, France (Meyer); Service de Médecine Interne, Centre Hospitalier Universitaire Hôtel Dieu De France, Beirut, Lebanon (Tohmé); Laboratoire Immunochimie, Assistance Publique–Hôpitaux de Paris, Groupe Hospitalier Pitié-Salpêtrière, Paris, France (Charuel, Musset).