Un jeune adulte atteint d’une maladie génétique grave du sang, la β-thalassémie, a été traité avec succès grâce à un protocole de thérapie génique utilisant un nouveau vecteur. Cette thérapie génique s’avère suffisamment efficace pour que, plus de trois ans après le début du traitement, le patient n’ait plus besoin de transfusions sanguines pour mener une existence normale alors qu’il était jusqu’alors transfusé tous les mois pour permettre sa survie. Les résultats de cet essai clinique, dirigé par le Pr. Philippe Leboulch et mené par des équipes de recherche associant le CEA (1), l’Inserm, l’Assistance Publique-Hôpitaux de Paris, l’Université Paris-Sud 11, les universités Paris Descartes et Paris Diderot (Sorbonne Paris Cité), les universités américaines d’Harvard et de Pennsylvanie, et la société Bluebird Bio (anciennement Genetix Pharmaceuticals) dont la filiale française est promotrice de l’essai, sont publiés dans la revue Nature du 16 septembre.
Les investigateurs principaux cliniques sont le Pr. Marina Cavazzana-Calvo de l’hôpital Necker-Enfants malades (AP-HP, Université Paris Descartes) et le Pr. Eliane Gluckman de l’hôpital Saint-Louis (AP-HP, Université Paris Diderot) à Paris. Ce premier succès concrétise les espoirs placés dans l’utilisation de la thérapie génique pour traiter les hémopathies. C’est aussi la première fois qu’une thérapie génique efficace est développée pour une maladie génétique aussi fréquente.
Qu’est-ce que la β-thalassémie ?
Les hémoglobinopathies (β-thalassémie et drépanocytose) sont les maladies génétiques héréditaires les plus fréquentes au monde. Elles sont dues à des défauts innés du gène codant pour la β-globine, un constituant essentiel de la molécule d’hémoglobine qui transporte l’oxygène dans les globules rouges, depuis les poumons vers tous les tissus de l’organisme. Dans les formes sévères de β-thalassémie, les patients souffrent d’anémie profonde. Leur survie est assurée par des transfusions régulières et l’élimination systématique du fer (chélation [2]). La présence de ce dernier à haute dose, très toxique, est due aux nombreuses transfusions. Les malades présentent des épaississements osseux et une augmentation du volume de la rate qui peut nécessiter son ablation. Près de 5 % de la population mondiale est porteur d’un gène de globine défectueux, près de 200 000 enfants naissent chaque année dans le monde avec un syndrome thalassémique, et 1 personne sur 10 000 est diagnostiquée avec cette maladie chaque année au sein de l’Union européenne. Dans la variété de thalassémie βE/b0, comme dans le cas du patient traité dans cet essai clinique, l’un des gènes de β-globine est complètement silencieux alors que l’autre exprime un taux faible d’une protéine mutée (βE). Cette variété est particulièrement fréquente en Asie du Sud Est et dans les populations immigrantes en provenance de ces pays.
Comment est-elle traitée actuellement ?
Le traitement conventionnel de la thalassémie majeure est basé sur des transfusions sanguines, généralement toutes les 3 ou 4 semaines, dès que le taux d’hémoglobine descend en dessous d’une valeur compatible avec une activité normale du patient. En l’absence de ces transfusions le patient succomberait rapidement d’anémie profonde. Mais leur caractère répété apporte un excès de fer qui doit être éliminé par un traitement chélateur, afin d’éviter des effets secondaires graves et parfois mortels. Si ce traitement a transformé l’espérance de vie des patients, sa lourdeur altère fortement leur qualité de vie. Quand c’est possible, une greffe de moelle osseuse peut être envisagée chez les enfants et les adolescents, mais ce traitement reste soumis à la difficulté de trouver un donneur compatible et à un risque substantiel de rejet de greffe ou de réaction du greffon contre l’hôte.
Production de β-globine par thérapie génique : un véritable challenge technique
La mise au point d’une thérapie génique est donc un véritable espoir pour les 80 % de malades ne pouvant pas avoir accès à une greffe de moelle. Envisagée depuis près de trente ans, cette thérapie génique représente en réalité un véritable défi technologique. La première étape a consisté à mettre au point le protocole de thérapie génique chez l’animal. Plusieurs obstacles majeurs ont dû être levés, ceci grâce à une étroite coopération entre la recherche « académique » dirigée par le Pr. Leboulch au MIT (Massachusetts Institute of Technology) puis à l’Université d’Harvard, en collaboration avec ses partenaires français et la société de biotechnologie américaine, qu’il a fondée, Bluebird Bio.
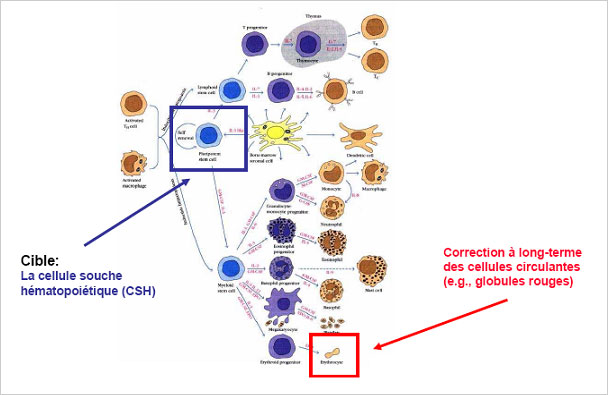
Prérequis de la thérapie génique hématopoïétique par « addition de gène » © CEA
En effet, le gène modifié de la β-globine à transférer est de grande taille. De plus, il faut qu’il soit intégré dans une grande proportion de cellules souches hématopoïétiques(3) et exprimé fortement et spécifiquement dans les globules rouges. Afin de permettre d’atteindre un niveau d’expression optimal, les chercheurs se sont tournés vers les vecteurs dérivés du virus d’immunodéficience humaine inactivé, particulièrement efficaces pour transférer de grands fragments d’ADN dans les cellules souches hématopoïétiques. Ces mêmes vecteurs ont également été utilisés récemment pour traiter avec succès deux patients atteints d’adrénoleucodystrophie, essais auxquels les Pr. Hacein-Bey-Abina, Cavazzana-Calvo et Leboulch ont aussi participé (Cartier et al.Science 2009;326:818-23).
Ces vecteurs sont dotés d’éléments nécessaires pour assurer une très forte expression de la β-globine. Ils comportent également des éléments de sécurisation qui évitent d’une part la réplication du virus et d’autre part l’activation non désirée de gènes du patient autres que la β-globine.
Le groupe du Pr. Leboulch avait ainsi réalisé, il y a près de 10 ans, la première correction de la drépanocytose chez la souris (Pawliuk et al.Science 2001;294:2368-71) avant d’appliquer ce protocole aux cellules souches hématopoïétiques humaines.
La deuxième étape du processus a consisté à produire à grande échelle des lots de vecteurs pour la thérapie de qualité « clinique ». Cette étape cruciale pour permettre l’application du protocole chez l’homme dans des conditions de sécurité optimale pour les patients a été prise en charge par la société Bluebird Bio.
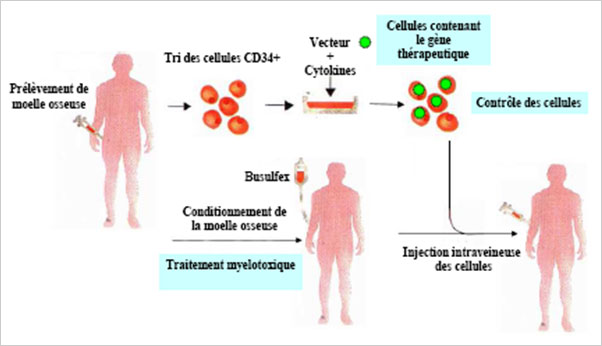
Premier essai clinique utilisant un vecteur lentiviral © CEA
L’essai clinique et ses résultats
Après obtention de l’autorisation des autorités sanitaires françaises (Afssaps) en 2006, un essai clinique de phase I/II pour la thalassémie a été lancé sous la direction scientifique du Pr. Leboulch. Le premier patient thalassémique est entré dans le protocole en 2007. Ce jeune homme âgé à l’époque de 18 ans, d’origine Thaïlandaise et Vietnamienne, était dépendant depuis son enfance de transfusions mensuelles pour rester en vie.
L’étape importante de transduction des cellules a été réalisée dans le Département de Biothérapie de l’Hôpital Necker-Enfants Malades par le Pr. Marina Cavazzana-Calvo et le Pr. Salima Hacein-Bey-Abina qui possèdent une grande expérience dans ce domaine. Elle a consisté à prélever les propres cellules de moelle osseuse du patient et à les corriger ex vivo par transfert d’une version fonctionnelle du gène déficient à partir du vecteur produit par le laboratoire du Pr. Leboulch en collaboration avec bluebird bio. Les cellules ainsi corrigées ont ensuite été réinjectées au patient. L’essai clinique a ensuite été poursuivi à l’hôpital Saint-Louis sous la responsabilité clinique du Pr. Gluckman. Le Dr. Françoise Bernaudin de l’hôpital Intercommunal de Créteil a assuré le suivi du patient.
« L’insertion par thérapie génique d’une copie du gène de la b-globine a permis, au bout de quelques mois, aux cellules d’exprimer la b-globine thérapeutique à un taux suffisamment élevé pour que le patient n’ait eu besoin d’aucune transfusion depuis plus de deux ans ! C’est une avancée historique dans le combat contre cette maladie », explique le Pr. Cavazzana-Calvo, qui sera pour la suite de l’essai l’investigateur principal clinique.
Le Dr. Bernaudin qui a suivi le patient depuis sa petite enfance a ajouté « C’est merveilleux de voir que la vie de ce jeune homme a changé de cette manière : il est maintenant libre de transfusions et de chélation du fer par perfusions, et a un emploi en CDI à plein temps dans un restaurant. Nous arrivons même à lui faire des saignées régulièrement pour accélérer l’élimination du fer toxique qui s’était accumulé depuis des années à cause des transfusions. »
A la vue de ces résultats particulièrement prometteurs, en janvier 2010, l’Afssaps a autorisé l’inclusion d’un nouveau patient et la poursuite de l’essai. L’Association Française contre les Myopathies (AFM), qui a déjà contribué à ce projet grâce aux dons du Téléthon, va collaborer avec le promoteur Bluebird Bio pour la poursuite des essais cliniques sur les maladies génétiques de la globine et pour le développement de ces thérapies au service des patients. En parallèle, un accord de collaboration a été signé en novembre 2009 avec la Thaïlande pour étendre l’essai clinique à ce pays pour lequel la β-thalassémie représente un véritable problème de santé publique avec plus de 3 000 naissances par an d’enfants atteints de cette maladie. La société Bluebird Bio envisage aussi à brève échéance l’extension de l’essai aux états-Unis, où d’autres groupes de recherche académiques sont en passe de débuter leurs propres essais cliniques de thérapie génique lentivirale pour le traitement des hémoglobinopathies.
Dans le suivi post-thérapie génique, une analyse génomique très pointue a été effectuée en collaboration avec le Pr. Frédéric Bushman de l’Université de Pennsylvanie à Philadelphie pour s’assurer de l’absence de risque à long terme lié à l’insertion du gène thérapeutique dans l’ADN du patient. « Nous avons ainsi observé que parmi les clones (4) cellulaires génétiquement modifiés, l’un d’entre eux est dominant (HMGA2), produisant environ 3 % des cellules nucléées sanguines. Ce clone reste stable depuis sa découverte il y a un an. Même s’il faut rester prudent, à ce jour ce clone reste sans conséquence hématologique délétère » souligne le Dr. Emmanuel Payen, chercheur Inserm dans le laboratoire du Pr. Leboulch.
(1) L’institut des maladies émergentes et des thérapies innovantes du Commissariat à l’énergie atomique et aux énergies alternatives (CEA) et son unité mixte de recherche (UMR 962) Inserm-CEA-Université Paris-Sud 11
(2) Chélation : utilisation d’une molécule capable de fixer le fer en surplus et ainsi de l’éliminer de l’organisme
(3) Cellules souches à la base de la production de toutes les cellules sanguines
(4) Clone cellulaire : ensemble de cellules génétiquement identiques dérivant d’une seule cellule initiale par divisions successives
Ces contenus pourraient aussi vous intéresser :
Contacts
Sources
"Transfusion independence and HMGA2 activation after gene therapy of human β-thalassemia"Marina Cavazzana-Calvo, Emmanuel Payen, Olivier Negre, Gary Wang, Kathleen Hehir, Floriane Fusil, Julian Down, Maria Denaro, Troy Brady, Karen Westerman, Resy Cavallesco, Beatrix Gillet-Legrand, Laure Caccavelli, Riccardo Sgarra, Leila Maouche-Chrétien, Françoise Bernaudin, Robert Girot, Ronald Dorazio, Geert-Jan Mulder, Axel Polack, Arthur Bank, Jean Soulier, Jérôme Larghero, Nabil Kabbara, Bruno Dalle, Bernard Gourmel, Gérard Socié, Stany Chrétien, Nathalie Cartier, Patrick Aubourg, Alain Fischer, Kenneth Cornetta, Frédéric Galacteros, Yves Beuzard, Eliane Gluckman, Frederick Bushman, Salima Hacein-Bey-Abina & Philippe LeboulchNature, 2010, Online (numéro du 16 septembre)