Contact Chercheur
Luis Garcia Responsable de l’équipe “Biothérapies des Maladies Neuromusculaires” - CNRS+ 33 (0)1 70 42 94 16 / + 33 (0)6 07 29 14 32
rf.qsvu@aicrag.siul
Des travaux de recherche associant des chercheurs du CNRS, de l’UVSQ et de l’Inserm au sein du laboratoire END-ICAP[1], en collaboration avec une équipe de l’université de Berne, démontrent le potentiel thérapeutique d’une nouvelle classe d’oligonucléotides[2] de synthèse pour le traitement de la myopathie de Duchenne (DMD) par chirurgie de l’ARN. Testée chez la souris, cette nouvelle génération de molécules se révèle cliniquement supérieure à celles en cours d’évaluation chez les patients DMD, notamment au niveau des fonctions cardiaque et respiratoire et du système nerveux central. Ces résultats sont publiés le 2 février 2015 dans la revue Nature Medecine.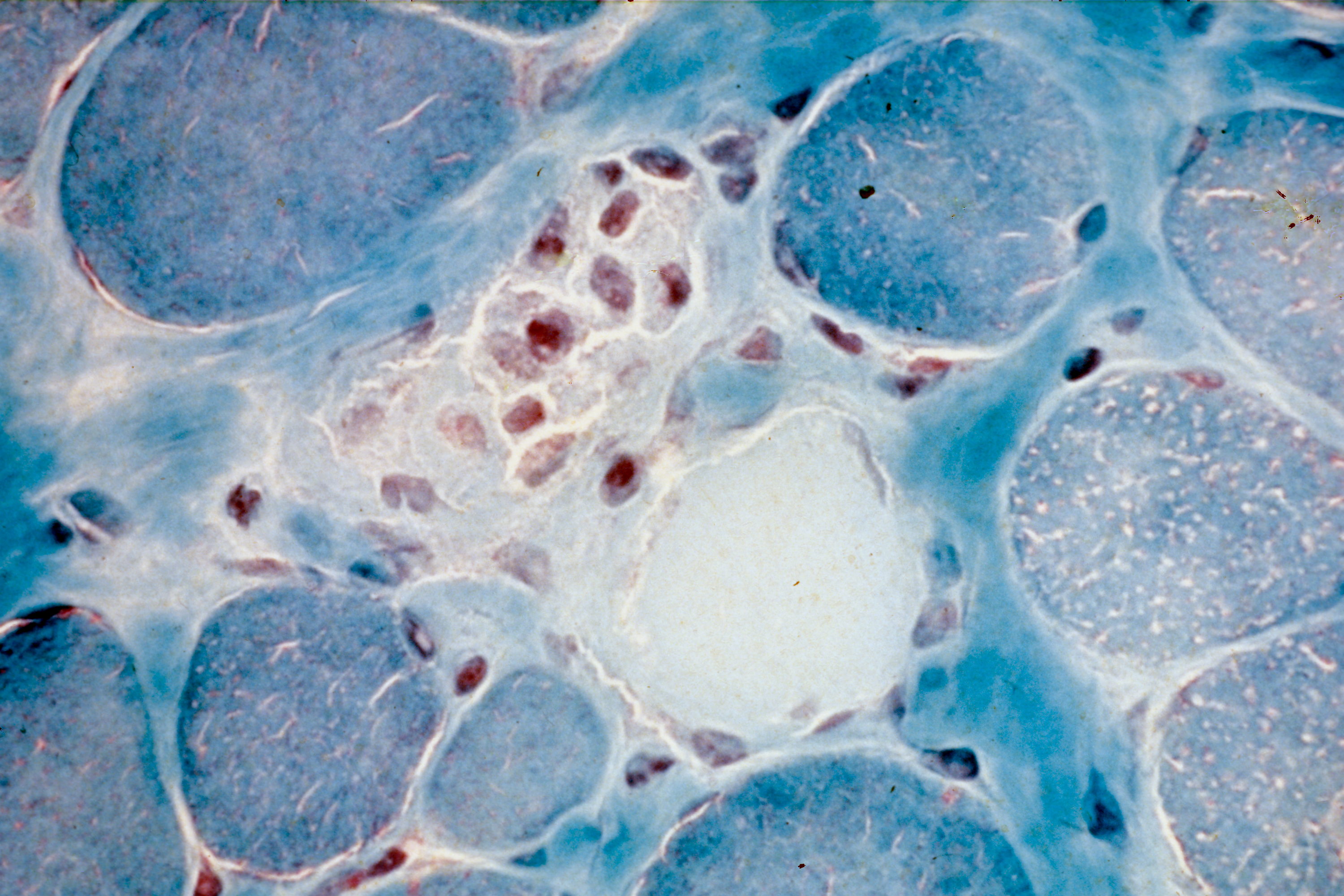
Etude de la myopathie de Duchenne – Lésions nécrotiques (mort cellulaire) de fibres musculaires constituées de myofibrilles. Coloration trichrome de Gomori. ©Inserm/Fardeau, Michel
Les maladies neuromusculaires regroupent un ensemble de plusieurs centaines de maladies, principalement d’origine génétique, définies par un défaut de commande du muscle ou par une destruction du tissu musculaire. Conjointement, elles affectent plusieurs dizaines de milliers de personnes en France et constituent un enjeu majeur de santé publique. La plus emblématique d’entre elles, la dystrophie musculaire de Duchenne (DMD) est causée par des mutations qui affectent le gène codant pour la dystrophine, une protéine indispensable au bon fonctionnement des cellules musculaires. Cette myopathie particulièrement sévère et très invalidante ne bénéficie encore d’aucun traitement satisfaisant.
La « chirurgie » de l’ARN est une approche développée dans le but de corriger certaines anomalies génétiques. Cette thérapie est fondée sur l’utilisation de petites séquences d’oligonucléotides antisens (AON)[3] capables de s’hybrider spécifiquement avec des ARN messagers, d’agir sur ces ARN et de permettre la synthèse d’une protéine manquante. Plusieurs études sont en cours pour synthétiser différents types d’AON destinés à agir sur la production de dystrophine. Malgré les résultats encourageants de certains essais cliniques, ces AON existants présentent des limites : leur niveau de toxicité reste parfois élevé et ils ne peuvent pas agir au niveau cardiaque ou passer la barrière hémato-encéphalique. La conception d’un traitement efficace simultanément pour l’ensemble de la musculature squelettique, le cœur et le système nerveux central reste encore un défi.
Les auteurs de ces travaux ont mis au point de nouveaux nucléotides pour la synthèse des AON : les tricyclo-DNA (tcDNA). Les AON-tcDNA, analogues synthétiques de l’ADN, s’hybrident avec les ARN cibles et vont entraîner l’excision d’un fragment de l’ARN[4]. En agissant ainsi sur la partie du gène comportant une erreur, ils permettent la synthèse d’une dystrophine certes tronquée mais stable et fonctionnelle. Le suivi chez les souris DMD traitées par ces AON-tcDNA montre qu’ils sont plus performants que leurs équivalents des générations précédentes. Administrés par voie intraveineuse, ils sont distribués efficacement à l’ensemble de la musculature squelettique. Ils atteignent aussi le tissu cardiaque et accèdent au système nerveux central, ce qui n’était pas le cas de leurs prédécesseurs. La restauration de la production de dystrophine est également plus efficace qu’avec les AON précédents. Après une douzaine de semaines de traitement hebdomadaire, les souris présentent une amélioration très significative de la fonction musculaire et surtout des fonctions respiratoire et cardiaque, qui sont les principales cibles à atteindre chez les patients souffrant de cette myopathie.
Les chercheurs ont aussi mis en évidence une correction des réponses émotionnelles naturellement exacerbées chez les sujets dystrophiques et pouvant entraîner des retards d’apprentissage et des défauts cognitifs. Cette partie de l’étude, menée en collaboration avec une équipe de l’Institut des neurosciences Paris Saclay (CNRS/Université Paris-Sud), démontre que la dystrophine est cruciale pour le bon fonctionnement de certains neurones et que les problèmes comportementaux observés lorsqu’il y a un déficit de cette protéine sont au moins partiellement réversibles chez la souris dystrophique adulte.
En plus de ces résultats prometteurs, les AON-tcDNA sont caractérisés par un temps long de persistance au sein des tissus ce qui permettrait à terme d’espacer les traitements. Autre avantage, ils ne sont pas dégradés mais évacués progressivement par l’organisme, permettant ainsi la réversibilité du traitement et limitant sa toxicité. Les analyses toxicologiques nécessaires sont toujours en cours mais les premiers résultats semblent en effet indiquer que ces nouveaux AON sont bien tolérés à fortes doses chez la souris.
Les mécanismes responsables de l’efficacité de ces AONs de troisième génération sont encore mal compris mais plusieurs de leurs propriétés pourraient entrer en jeu, notamment leur forte affinité pour l’ARN et leur capacité à former spontanément des agrégats de type « nanoparticules ».
Ces travaux s’inscrivent dans le cadre d’un vaste projet collaboratif international (ICE – International Collaborative Effort for DMD) à l’initiative de l’Association monégasque contre les myopathies (AMM) et du Duchenne Parent Project France (DPP-F) et sont pour partie soutenus par la Chaire d’excellence HandiMedEx – Investissements d’avenir.
[2] Les oligonucléotides sont des courts segments d’acides nucléiques (ARN ou ADN).
[3] Ces séquences sont appelées antisens car elles sont complémentaires de l’ARN messager. Le brin de synthèse aura donc une séquence inverse de celle du brin d’ARN.
[4] Après la transcription de l’ADN en ARN, l’ARN va subir un certain nombre de modifications, dont l’épissage, au cours duquel des fragments non-codants vont être exclus afin de donner l’ARN mature utilisé pour la traduction en protéines.
[5] Administré par voie générale.