
Crédits: C Kowalski/Université Paris-Descartes
Première médicale : Syndrome de CLOVES et syndromes d’hypercroissance : amélioration remarquable de l’état de santé de 19 patients enfants et adultes grâce à une nouvelle stratégie thérapeutique
Le Dr Guillaume Canaud de l’hôpital Necker-Enfants malades – AP-HP, l’Université Paris Descartes, l’Inserm (INEM l’Institut Necker Enfants Malades – Centre de médecine moléculaire) et son équipe viennent de démontrer l’efficacité d’un nouveau médicament, un inhibiteur spécifique appelé BYL719, dans une cohorte de 19 patients suivis à l’hôpital Necker-Enfants Malades – AP-HP et souffrant du syndrome de CLOVES (Congenital Lipomatous Overgrowth, Vascular Malformation, Epidermal Nævi) ou de troubles apparentés. Ce médicament est actuellement en cours d’essai thérapeutique en cancérologie (phase I/II). Aucun effet secondaire significatif n’a été constaté 18 mois après le début du traitement. Cette étude, publiée dans la revue Nature, représente un exemple de médecine de précision et démontre l’intérêt majeur de cette stratégie thérapeutique pour ces patients qui voient leur état de santé et leur qualité de vie s’améliorer de manière significative.
Les patients souffrant du syndrome de CLOVES (Congenital Lipomatous Overgrowth, Vascular Malformation, Epidermal Nævi) ou de troubles apparentés présentent des déformations majeures et des tuméfactions vasculaires dues à des mutations d’un gène, appelé PIK3CA. Ce gène régule la prolifération et la croissance des cellules. Lorsqu’il est trop activé il est responsable de croissance excessive des parties du corps touchées par la mutation. Ainsi la présentation clinique des patients est très variable en fonction du nombre de tissus affectés pouvant aller d’une macrodactylie (gros doigt isolé) à des formes très sévères touchant l’ensemble du corps telles que le syndrome de CLOVES.
Au cours des formes les plus graves, il existe des excroissances de tissu graisseux, des malformations vasculaires, une scoliose, des manifestations touchant le squelette comme un élargissement majeur des os ou encore des déformations d’organes tel que le cerveau ou les reins. Jusqu’à présent aucun traitement curatif n’était disponible pour ces patients dont le pronostic pouvait être engagé à court ou moyen terme et pour lesquels, les seules options thérapeutiques consistaient en des traitements symptomatiques, et pour les cas les plus graves, à subir des embolisations ou des chirurgies mutilantes pour préserver les organes ou les membres sains. Enfin, il est important de noter que ces syndromes sont fréquemment associés à des douleurs chroniques et ont un retentissement majeur sur la qualité de vie des patients et leur vie sociale.
Le gène PIK3CA est fréquemment muté dans un certain nombre de cancers (sein et colon notamment) et constitue une cible thérapeutique pour l’industrie pharmaceutique. Les mutations de PIK3CA dans les cancers sont les mêmes que celles retrouvées chez les patients atteints de syndrome de CLOVES et troubles apparentés.
Fin 2015, le Dr Guillaume Canaud, spécialiste de cette voie moléculaire, a été confronté à un patient de 29 ans porteur d’un syndrome de CLOVES très évolué avec un pronostic engagé pour lequel plus aucune chirurgie ou embolisation radiologique ne pouvait être proposée. le Dr Guillaume Canaud s’est alors rapproché du laboratoire Novartis qui travaille au développement en cancérologie d’un inhibiteur spécifique du gène PIK3CA appelé BYL719. Ce médicament est actuellement en cours d’essai thérapeutique en oncologie (phase I/II).
En janvier 2016, après avoir obtenu l’autorisation de l’ANSM d’utiliser ce médicament expérimental, le Dr Guillaume Canaud a démarré le traitement chez ce premier patient. Très rapidement, un effet positif a été observé sur l’ensemble des symptômes. Il a notamment été constaté une diminution importante des masses vasculaires et des excroissances dont le patient souffrait mais aussi une amélioration majeure de sa qualité de vie. Dix-huit mois plus tard, ce premier patient n’a présenté qu’un seul effet secondaire, une hyperglycémie, bien contrôlée par un simple régime alimentaire.
En parallèle, afin de mieux comprendre cette pathologie et le mode de fonctionnement du médicament, le Dr Canaud a créé avec son équipe de recherche au sein de l’INEM-Unité Inserm U1151), le premier modèle de souris (modèle murin) au monde regroupant l’ensemble des lésions dont souffrent les patients. Les souris ont été traitées avec le médicament BYL719 et là encore une amélioration majeure et rapide de leur état a pu être constatée.
Fort de ces résultats, le Dr Canaud a rapidement constitué en juin 2016, un groupe de travail réunissant une dizaine de spécialités médicales et chirurgicales prenant en charge les patients atteints de syndrome de CLOVES ou apparentés au sein de l’hôpital Necker – Enfants malades – AP-HP. L’idée de ce groupe était de mieux prendre en charge ces patients.
Au cours de l’été 2016, une enfant de 9 ans atteinte d’une forme sévère du syndrome de CLOVES avec une tuméfaction vasculaire menaçant sa vie et pour laquelle un acte chirurgical ou d’embolisation n’était pas possible, a bénéficié de ce traitement expérimental. De nouveau, le BYL719 a eu un effet spectaculaire sur l’ensemble des symptômes, déformations et sur la tuméfaction vasculaire. Il est important de noter qu’aucun effet secondaire n’a été constaté chez ce premier enfant, le premier dans le monde à recevoir ce traitement, et que sa croissance n’a pas été affectée au cours des 12 mois du suivi.
En février 2017, 17 nouveaux patients (14 enfants et 3 adultes âgés de 4 ans à 50 ans) suivis à l’hôpital, et pour lesquels le pronostic était engagé ou une chirurgie mutilante programmée, ont bénéficié, grâce à une autorisation de l’ANSM, du traitement par BYL719 fourni par Novartis. Dès les premiers jours après initiation du traitement, tous les patients ont présenté une amélioration spectaculaire de leur état général et notamment une réduction rapide de la taille des tumeurs vasculaires, des dilatations veineuses, de l’aspect cutané ou du volume anormal des membres ainsi qu’une diminution de la fatigue et une meilleure résistance à l’effort. Par ailleurs, tous les patients ont connu une amélioration de leur scoliose. Ils ont ainsi pu selon les cas reprendre une activité physique, arrêter les traitements à base de morphine, retourner à l’école, …
Après six mois de traitement, ces 17 patients sont encore en vie et aucune intervention chirurgicale n’a été effectuée. Des effets secondaires mineurs, tels que des aphtes, ont été observés chez trois d’entre eux.
Les 19 patients continuent de recevoir quotidiennement le BYL719.
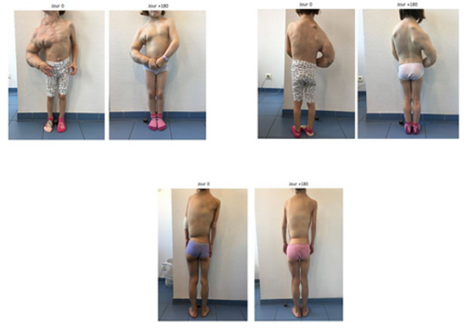
Patients à j 0 et à j + 180 – © Dr Canaud, AP-HP
Cette étude, dont le protocole a été approuvé par l’Agence Nationale de Sécurité du Médicament et des Produits de Santé (ANSM), démontre l’efficacité de cette approche thérapeutique.
Pour le Dr Canaud, « ce traitement va radicalement changer le devenir des patients porteurs de syndromes d’hypercroissance associés à une mutation de PIK3CA. Le médicament a permis d’obtenir des résultats dépassant nos espérances avec des régressions de malformations, pourtant présentes depuis de nombreuses années, mais aussi une amélioration de la qualité de vie des patients et de leur entourage. Le BYL719 représente ainsi un formidable espoir thérapeutique même pour des formes très sévères. Enfin, notre étude démontre l’intérêt de mettre au point des traitements ciblés dans les maladies génétiques pour développer une médecine dite de précision, mais également la nécessité d’une très forte interaction entre cliniciens et chercheurs pour faire avancer la connaissance et le développement de nouveaux médicaments. »
Ces travaux ont fait l’objet du dépôt d’une demande de brevet par Inserm Transfert au nom de l’AP-HP, de l’Université Paris Descartes et de l’Inserm.
Ces contenus pourraient aussi vous intéresser :

Contacts
Sources
Molecular targeted therapy in patients with PIK3CA-related overgrowth syndromeQuitterie Venot1, Thomas Blanc1,2,3*, Smail Hadj Rabia2,4,5*, Laureline Berteloot5,6, Sophia Ladraa1, Jean-Paul Duong2,7, Estelle Blanc8, Simon C. Johnson9, Clément Hoguin1, Olivia Boccara4, Sabine Sarnacki2,3, Nathalie Boddaert2,5,6, Stephanie Pannier2,10, Frank Martinez11, Sato Magassa1, Junna Yamaguchi1, Bertrand Knebelmann1,2,11, Pierre Merville12,13, Nicolas Grenier14, Dominique Joly1,2,11,Valérie Cormier-Daire2,5,15, Caroline Michot2,5,15, Christine Bole-Feysot5, Arnaud Picard2,16, Véronique Soupre16, Stanislas Lyonnet2,5,15, Jeremy Sadoine17, Lotfi Slimani17, Catherine Chaussain2,17, Cécile Laroche-Raynaud18, Laurent Guibaud19, Christine Broissand20, Jeanne Amiel2,5,15, Christophe Legendre1,2,11, Fabiola Terzi1,2, Guillaume Canaud1,2,11†.Nature, 13 juin 2018https://doi.org/10.1038/s41586-018-0217-9