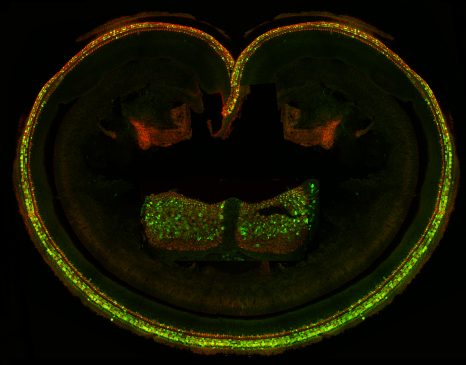
Organe de Corti et crête ampullaire du vestibule (insert) d’oreille interne injectée avec le virus AAV8 produisant la GFP et la protéine sans. Les cellules sensorielles cochléaires et vestibulaires ont été immunomarquées pour la GFP et la myosine VI, et analysées au microscope confocal. Les cellules vertes et orange produisent la protéine du gène thérapeutique.
© Institut Pasteur
Des chercheurs de l’Institut Pasteur, de l’Inserm et du CNRS* viennent de restaurer, pour la première fois, l’audition et l’équilibre dans un modèle murin du syndrome de Usher de type 1G (USH1G). Grâce à l’injection locale du gène USH1G, essentiel pour la formation et le maintien de l’appareil de transduction mécano-électrique des cellules sensorielles de l’oreille interne, les chercheurs ont réussi à rétablir le fonctionnement de cette structure, et ont ainsi permis à un modèle murin de ce syndrome, de recouvrer l’ouïe et l’équilibre. Ces résultats, publiés dans la revue PNAS, ouvrent la voie vers le développement de traitements, par thérapie génique, de certaines formes génétiques de surdité.
La surdité, associée dans certains cas à des troubles de l’équilibre, est le déficit sensoriel le plus fréquent. Elle affecte plus de 280 millions de personnes dans le monde, selon l’OMS. En France, 1 enfant sur 700 naît avec une surdité sévère ou profonde, et 1 enfant sur 1000 deviendra malentendant avant l’âge adulte.
Depuis 20 ans, des progrès considérables ont été réalisés dans la compréhension des surdités héréditaires présentes dès la naissance, dont la cause la plus fréquente est un dysfonctionnement de l’oreille interne. L’oreille interne est constituée de l’organe de l’audition (cochlée) et des cinq organes de l’équilibration (saccule, utricule, et trois canaux semi-circulaires), contenant les cellules sensorielles ou cellules ciliées. A ce jour, près de 100 gènes responsables de ces surdités ont été identifiés.
Parmi les différentes formes génétiques de surdité, le syndrome de Usher de type 1 (USH1) est caractérisé par une surdité congénitale profonde, des troubles de l’équilibration, et une atteinte visuelle progressive qui évolue vers la cécité. Ce syndrome peut être causé par des mutations dans 5 gènes différents, dont le gène USH1G codant pour une protéine « d’échafaudage » nécessaire à la cohésion de la touffe ciliaire des cellules ciliées.
Actuellement, les individus atteints de surdité et de troubles de l’équilibre sont équipés de prothèses auditives et peuvent bénéficier d’une rééducation pour améliorer leurs troubles de l’équilibre, mais les résultats sont variables. Une alternative envisageable pour traiter les surdités d’origine génétique est la thérapie génique, c’est-à-dire le transfert d’une copie saine (non mutée) du gène défectueux, afin de rétablir l’expression de la protéine déficiente. Cependant, à ce jour, seule une amélioration partielle de l’audition a pu être obtenue dans des modèles murins de formes particulières de surdité humaine, qui ne comportaient pas d’anomalie sévère de la structure des cellules ciliées.
Dans ce contexte, des chercheurs de l’Institut Pasteur, de l’Inserm et du CNRS* viennent de restaurer l’audition et l’équilibre chez un modèle murin du syndrome USH1 grâce à une thérapie génique. Par une injection locale unique, après la naissance, du gène USH1G, les chercheurs ont réussi à rétablir la structure, très endommagée dès la naissance, de l’appareil de transduction mécano-électrique des cellules ciliées, et ont ainsi permis aux souriceaux de recouvrer, et ce de manière durable, partiellement l’ouïe et complètement l’équilibre.

Touffes ciliaires de cellules sensorielles vestibulaires analysées au microscope électronique à balayage. On peut distinguer, une touffe ciliaire normale avec sa forme caractéristique agencée en « forme d’escalier » (couleur jaune), une touffe ciliaire défectueuse Usher1g (en rose), et une touffe ciliaire Usher1g traitée (en vert) dont la forme normale/caractéristique à été restaurée par la thérapie génique. © Institut Pasteur
Les chercheurs ont procédé à l’injection du gène USH1G dans l’oreille interne en utilisant le virus AAV8, inoffensif pour la santé mais permettant de cibler spécifiquement les cellules ciliées. L’expression du gène médicament a été détectée dès 48 heures après l’injection. Les chercheurs ont démontré qu’une seule injection, en rétablissant la production et la localisation de la protéine concernée dans les cellules ciliées, est suffisante pour améliorer l’audition et l’équilibration chez les souriceaux atteints. Ces résultats suggèrent que la protéine médicament a pu interagir normalement avec ses partenaires de liaison au sein du complexe moléculaire USH1 (c’est-à-dire avec les protéines cadhérine 23, protocadhérine 15, myosine VIIa, et harmonine), comme requis pour le bon fonctionnement des canaux de la transduction mécano-électrique.
Comme l’explique Saaïd Safieddine, directeur de recherche du CNRS à l’Institut Pasteur et dernier auteur de l’étude, « nous venons de prouver qu’il est possible de corriger partiellement une forme génétique particulière de surdité accompagnée de troubles de l’équilibre, grâce à une thérapie génique locale effectuée après le stade du développement de l’oreille qui est affecté le premier par la mutation responsable. La fenêtre de temps pour traiter efficacement le syndrome USH1 par thérapie génique pourrait donc être plus large qu’initialement envisagée. »
Cette étude constitue une étape importante vers la conception d’essais cliniques de thérapie génique en vue d’un traitement curatif de certaines formes génétiques de surdité chez l’Homme.
*Du laboratoire Génétique et physiopathologie de l’audition (Institut Pasteur/Inserm/UPMC), du laboratoire Gènes, synapses et cognition (CNRS/Institut Pasteur) et du Centre de neurophysique, physiologie, pathologie (CNRS/Université Paris-Descartes).
Ces contenus pourraient aussi vous intéresser :
Contacts
Sources
Local gene therapy durably restores vestibular function in a mouse model of Usher syndrome type 1G, PNAS, 5 septembre 2017 Alice Emptoz (a,b,c), Vincent Michela (b,c), Andrea Lellia (b,c), Omar Akil (d), Jacques Boutet de Monvela (b,c), Ghizlene Lahloua (b,c), Anaïs Meyera (b), Typhaine Duponta (b,c), Sylvie Nouaillea (b,c), Elody Ey €, Filipa Franca de Barros (f), Mathieu Beraneck (f), Didier Dulon (g), Jean-Pierre Hardelina (b,c), Lawrence Lustig (h), Paul Avan (i), Christine Petita (b,c,j), and Saaid Safieddine (a,b,c).
(a) INSERM, UMR 1120, Paris, France.
(b) Génétique et Physiologie de l’Audition, Institut Pasteur, 75015 Paris, France.
(c) Complexité du Vivant, Sorbonne Universités, Université Pierre-et-Marie-Curie, Université Paris VI, 75015 Paris, France.
(d) Otolaryngology-Head & Neck Surgery, University of California, San Francisco, CA 94117.
(e) Unité de Génétique Humaine et Fonctions Cognitives, Institut Pasteur, CNRS UMR 3571, 75015 Paris, France.
(f) Centre de Neurophysique, Physiologie, et Pathologie, CNRS UMR 8119, Université Paris-Descartes, 75006 Paris, France.
(g) Laboratoire de Neurophysiologie de la Synapse Auditive, Bordeaux Neurocampus, INSERM, UMR 1120, Université de Bordeaux, 33076 Bordeaux, France.
(h) Columbia University School of Medicine and New York Presbyterian Hospital, New York, NY 10034.
(i) Laboratoire de Biophysique Sensorielle, Faculté de Médecine, Université d’Auvergne, Biophysique Médicale, Centre Jean Perrin, 63000 Clermont-Ferrand, France.
(j) Collège de France, 75005 Paris, FranceDOI : 10.1073/pnas.1708894114