Une étude internationale de grande ampleur, impliquant des chercheurs français de l’Unité mixte de recherche Inserm-Institut Pasteur Lille-Université Lille Nord de France « Santé publique et épidémiologie moléculaire des maladies liées au vieillissement », dirigée par Philippe Amouyel, vient de mettre au jour un gène de susceptibilité d’une maladie rare à l’origine de la susceptibilité à une maladie fréquente, la maladie d’Alzheimer, témoignant ainsi de l’hétérogénéité de l’étiologie de la maladie d’Alzheimer. Cette approche de séquençage systématique des exons est détaillée dans un article paru dans la revue The New England Journal of Medicine datée du 14 novembre 2012.
L’ostéodysplasie polykystique lipomembraneuse avec leucoencéphalopathie sclérosante ou maladie de Nasu-Hakola est une affection génétique transmise sur le mode autosomique récessif. La maladie débute vers la trentaine par des douleurs du poignet ou de l’épaule associées à des gonflements articulaires. Des fractures osseuses surviennent pour des traumatismes mineurs. Les radiographies osseuses mettent en évidence des kystes épiphysaires. Apparaissent ensuite de légers changements de la personnalité suivi par des symptômes neurologiques frontaux (euphorie, perte de l’inhibition sociale) évoluant vers une démence à début précoce. Cette affection a été associée à des mutations dans le gène TREM2 (Triggering Receptor Expressed on Myeloid cells 2) sur le chromosome 6
Aujourd’hui, des chercheurs britanniques, américains et français, viennent de montrer que sur cette même région du chromosome 6, des mutations du gène TREM2 étaient associées à un risque 5 fois plus élevé de développer une maladie d’Alzheimer à début tardif. Un séquençage complet a été réalisé chez 281 individus avec une maladie d’Alzheimer et 504 témoins. L’analyse du gène TREM2 a permis de montrer un excès de mutations de TREM2 chez les malades par rapport aux témoins. La caractérisation d’une de ces mutations de TREM2, dans de très larges échantillons de populations de patients atteints de maladie d’Alzheimer a permis aux chercheurs de mesurer précisément la force importante de cette association entre mutation de TREM2 et la maladie. Enfin une étude de réplication a été réalisée dans une autre série indépendante de 1994 cas et 4602 contrôles qui est venue confirmer cette forte association (OR=4,97 IC95% [2,42-10,21], P<6.10-6).
Ces résultats sont aussi confirmés dans le même numéro de la revue The New England Journal of Medicine par une équipe islandaise qui montre également que ce gène est un facteur de risque de maladie d’Alzheimer dans la population finlandaise ainsi que dans d’autres populations européennes.
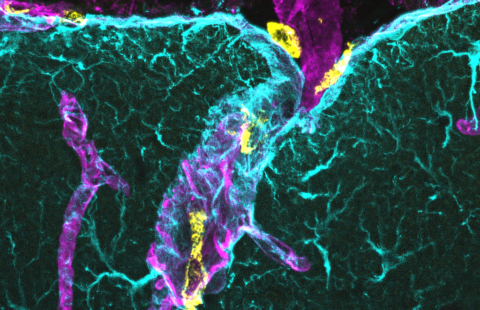
Une analyse anatomopathologique de six individus présentant des variants du gène TREM2 a mis en évidence des lésions cérébrales de type Alzheimer. L’étude de l’expression du gène TREM2 dans des cerveaux humains normaux a permis de montrer une localisation importante dans la substance blanche et dans l’hippocampe et le cortex. Dans un modèle de souris transgénique de la maladie d’Alzheimer, une augmentation d’expression de TREM2 a été observée dans les cellules microgliales entourant les plaques amyloïdes et les neurones comparativement à des souris normales. Le gène TREM2 code pour une protéine qui participe à l’activation de la réponse immunitaire dans les macrophages et les cellules dendritiques.
Cette découverte à deux conséquences principales. Tout d’abord cette observation permet de mieux comprendre l’implication du système immunitaire dans la maladie d’Alzheimer pour lequel le gène du récepteur du complément 1 (CR1) avait déjà été impliqué dans des travaux antérieurs de l’UMR744 Inserm-Lille2-IPL[1], ouvrant la voie à de nouvelles hypothèses de prise en charge de la maladie d’Alzheimer. Par ailleurs, cette approche de séquençage systématique des exons a permis de trouver un gène de susceptibilité d’une maladie rare à l’origine de la susceptibilité à une maladie fréquente, témoignant ainsi de l’hétérogénéité de l’étiologie de la maladie d’Alzheimer. C’est la perte d’activité de ce gène à l’état homozygote ou hétérozygote qui détermine la nature de l’affection.
Ces résultats qui témoignent des nombreuses avancées dans la compréhension de la maladie d’Alzheimer ont impliqué des équipes du labex Distalz, et ont pu être réalisés en partie grâce au soutien de la Fondation de Coopération Scientifique sur la maladie d’Alzheimer, qui coordonne le volet recherche du Plan de lutte contre la maladie d’Alzheimer et des maladies apparentées, lancé en février 2008.
Avec l’augmentation de la longévité des populations humaines, le nombre de patients atteints de maladie d’Alzheimer tend à augmenter en France et partout dans le monde. Première cause de troubles de la mémoire et des fonctions intellectuelles chez la personne âgée, cette affection constitue donc un enjeu majeur de santé publique.
La maladie d’Alzheimer est l’une des principales causes de dépendance de la personne âgée. Elle résulte d’une dégradation des neurones dans différentes régions du cerveau. Elle se manifeste par une altération croissante de la mémoire, des fonctions cognitives ainsi que par des troubles du comportement conduisant à une perte progressive d’autonomie. En France, la maladie d’Alzheimer touche plus de 850 000 personnes et représente un coût social et économique majeur.
La maladie d’Alzheimer est caractérisée par le développement dans le cerveau de deux types de lésions : les plaques amyloïdes et les dégénérescences neurofibrillaires. Les plaques amyloïdes proviennent de l’accumulation extracellulaire d’un peptide, le peptide β amyloïde (Aβ), dans des zones particulières du cerveau. Les dégénérescences neurofibrillaires sont des lésions intraneuronales provenant de l’agrégation anormale, sous forme de filaments, d’une protéine appelée protéine Tau.
L’identification des gènes qui participent à la survenue de la maladie d’Alzheimer et à son évolution permettra d’aborder plus rapidement les mécanismes physiopathologiques à l’origine de cette affection, d’identifier des protéines et des voies métaboliques cibles de nouveaux traitements et d’offrir des moyens d’identifier les sujets les plus à risque lorsque des traitements préventifs efficaces seront disponibles
[1] Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease.
Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, Combarros O, Zelenika D, Bullido MJ, Tavernier B, Letenneur L, Bettens K, Berr C, Pasquier F, Fiévet N, Barberger-Gateau P, Engelborghs S, De Deyn P, Mateo I, Franck A, Helisalmi S, Porcellini E, Hanon O; the European Alzheimer’s Disease Initiative Investigators, de Pancorbo MM, Lendon C, Dufouil C, Jaillard C, Leveillard T, Alvarez V, Bosco P, Mancuso M, Panza F, Nacmias B, Bossù P, Piccardi P, Annoni G, Seripa D, Galimberti D, Hannequin D, Licastro F, Soininen H, Ritchie K, Blanché H, Dartigues JF, Tzourio C, Gut I, Van Broeckhoven C, Alpérovitch A, Lathrop M, Amouyel P. Nature Genetics 2009. 41: 1094-1099.
Ces contenus pourraient aussi vous intéresser :
Contacts
Sources
“TREM2 Variants Predispose to Alzheimer’s Disease.” Rita Guerreiro, Ph.D., Aleksandra Wojtas, M.S., Jose Bras, Ph.D., Minerva Carrasquillo, Ph.D., Ekaterina Rogaeva, Ph.D., Elisa Majounie, Ph.D., Carlos Cruchaga, Ph.D., Celeste Sassi, M.D., John S.K. Kauwe, Ph.D., Steven Younkin, M.D., Ph.D., Lilinaz Hazrati, M.D., Ph.D., John Collinge, M.D., Jennifer Pocock, Ph.D., Tammaryn Lashley, Ph.D., Julie Williams, Ph.D., Jean-Charles Lambert, Ph.D., Philippe Amouyel, M.D., Ph.D., Alison Goate, Ph.D., Rosa Rademakers, Ph.D., Kevin Morgan, Ph.D., John Powell, Ph.D., Peter St. George-Hyslop, M.D., Andrew Singleton, Ph.D., and John Hardy, Ph.D. for the Alzheimer Genetic Analysis Group New England Journal of Medicine, 2012