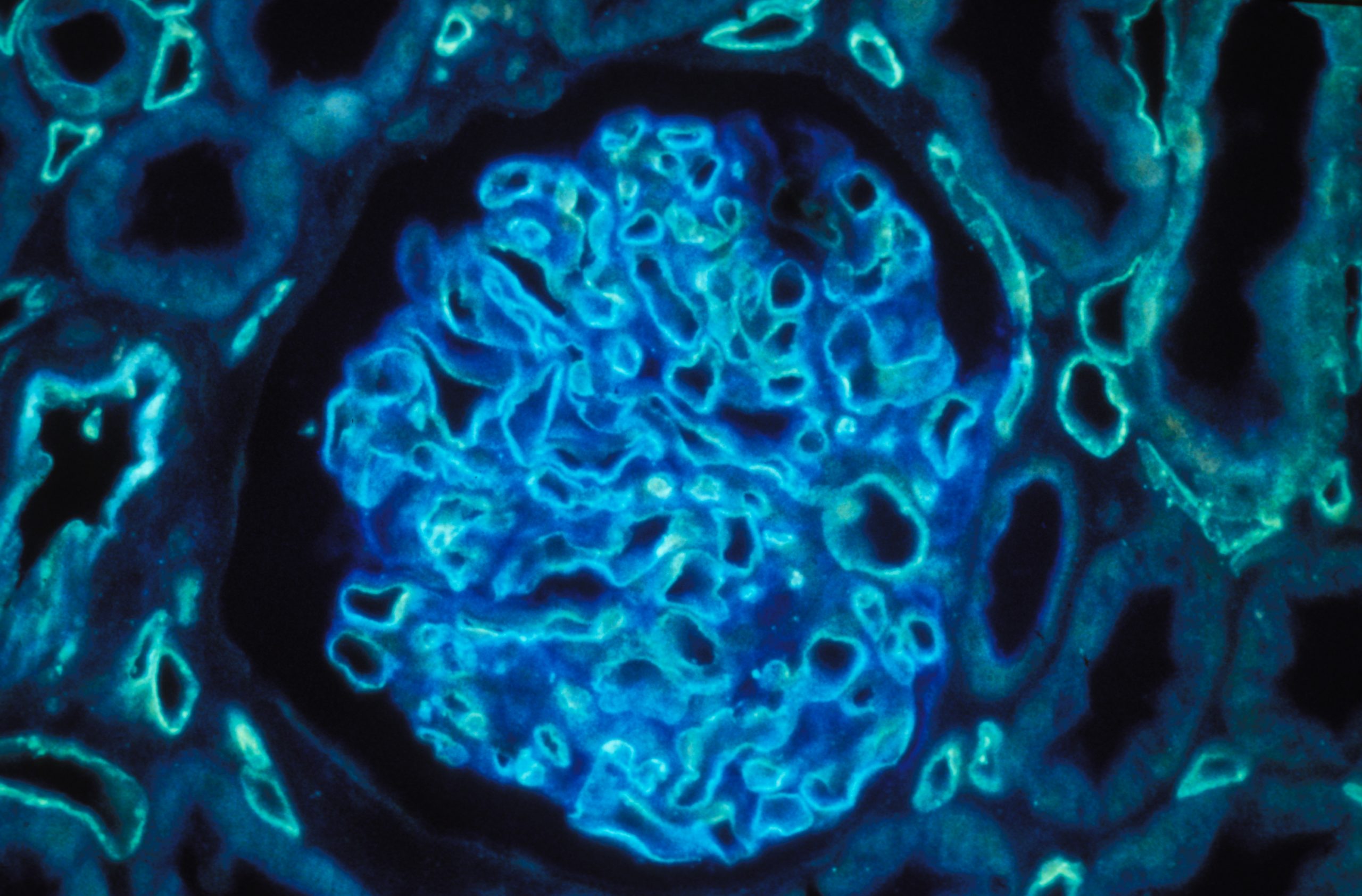
Coupe de rein humain grossie 400 fois par un microscope à immunofluorescence polychromatique.© Inserm/Oriol, Rafael
L’équipe composée de chercheurs et chercheuses du service d’endocrinologie et des maladies de la reproduction de l’hôpital Bicêtre AP-HP, de l’Inserm et de l’Université Paris-Saclay, a mené des travaux, coordonnés par le Professeur Peter Kamenický, pour étudier la cause génétique de l’hyperplasie bilatérale macronodulaire des surrénales avec syndrome de Cushing induit par l’alimentation. Cette maladie rare touche les deux glandes surrénales situées au-dessus des reins et entraine une surproduction du cortisol, une hormone stéroïde dont l’excès a des conséquences néfastes pour l’organisme. Les chercheurs ont pu déterminer l’explication moléculaire de la survenue de cette maladie 30 ans après sa description initiale. Ces travaux ont fait l’objet d’une publication le 13 octobre 2021 dans la revue The Lancet Diabetes & Endocrinology.
Cette forme rare du syndrome de Cushing surrénalien, étudiée par ces chercheurs, est due à l’expression anormale du récepteur du GIP (Glucose-dependent insulinotropic peptide), dans les deux glandes surrénales des patients. Le GIP est une hormone produite par l’intestin grêle en réponse à l’ingestion d’aliments. Chez les patients atteints de cette forme particulière du syndrome de Cushing, les concentrations de cortisol augmentent anormalement après chaque prise alimentaire. Les patients atteints de cette maladie développent les signes cliniques typiques du syndrome de Cushing tels que la prise de poids associée à une atrophie musculaire, l’hypertension artérielle, le diabète sucrée, l’ostéoporose et la dépression. La pathologie est associée à une augmentation de la mortalité, surtout des causes cardiovasculaires.
Dans cette étude internationale impliquant les chercheurs de six pays, et reposant notamment sur une collaboration étroite franco-québécoise, l’équipe rapporte que l’hyperplasie macronodulaire des surrénales GIP-dépendante, dans ses formes familiales comme sporadiques, est une maladie génétique, causée par des mutations germinales de Lysine Déméthylase 1A (KDM1A) avec une perte secondaire du second locus de KDM1A, comportant la seconde copie du gène, dans le tissu surrénalien. KDM1A agit principalement comme un répresseur transcriptionnel (i.e. un régulateur qui empêche un gène d’être exprimé), la perte de sa fonction aboutit à une dérégulation d’expression de différents gènes dans le tissu surrénalien, incluant le récepteur du GIP mais également d’autres récepteurs couplés aux protéines G.
Cette découverte permettra de proposer un conseil génétique et une détection plus précoce de cette maladie rare aux patients et à leurs apparentés. Les maladies rares sont en général sous-diagnostiquées. Ceci est d’autant plus important que les variations pathogènes de KDM1A prédisposent également au myélome et à d’autres types de cancer.
De plus, ce nouveau rôle de KDM1A comme régulateur épigénétique de l’expression du récepteur du GIP et d’autres récepteurs couplés aux protéines G pourrait avoir des implications pharmacologiques.
Ces contenus pourraient aussi vous intéresser :

Contacts
Sources
The Lancet Diabetes & Endocrinology.
Fanny Chasseloup, Isabelle Bourdeau, Antoine Tabarin, Daniela Regazzo, Charles Dumontet, Nataly Ladurelle, Lucie Tosca, Larbi Amazit, Alexis Proust, Raphael Scharfmann, Tiphaine Mignot, Frédéric Fiore, Stylianos Tsagarakis, Dimitra Vassiliadi, Dominique Maiter, Jacques Young, Anne-Lise Lecoq, Vianney Deméocq, Sylvie Salenave, Hervé Lefebvre, Lucie Cloix, Philippe Emy, Rachel Dessailloud, Delphine Vezzosi, Carla Scaroni, Mattia Barbot, Wouter de Herder, François Pattou, Martine Tétreault, Gilles Corbeil, Margot Dupeux, Benoit Lambert, Gérard Tachdjian, Anne Guiochon-Mantel, Isabelle Beau, Philippe Chanson, Say Viengchareun, André Lacroix, Jérôme Bouligand, Peter Kamenický.