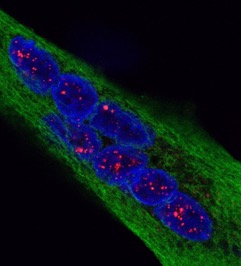
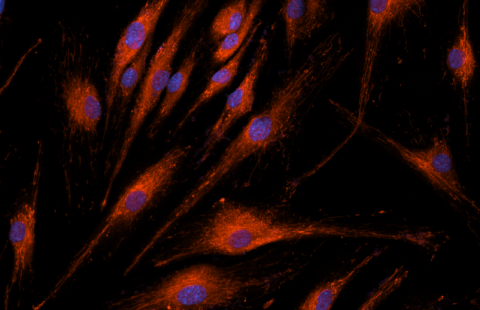
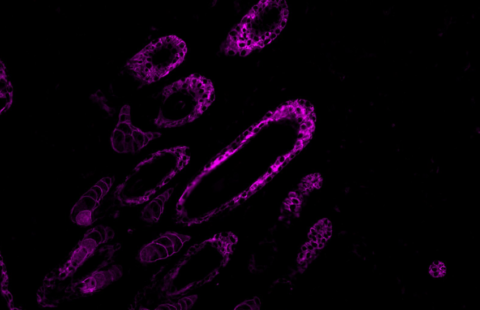
Reversal of RNA toxicity in myotonic dystrophy via a decoy RNA-binding protein with high affinity for expanded CUG repeats
Nature Biomedical Engineering, février 2022
DOI : https://doi.org/10.1038/s41551-021-00838-2
Ludovic Arandel1,&, Magdalena Matloka1,&, Arnaud F. Klein1, Frédérique Rau1, Alain Sureau1, Michel Ney1, Aurélien Cordier1, Maria Kondili1, Micaela Polay-Espinoza1, Naira Naouar1, Arnaud Ferry1,3, Mégane Lemaitre1,4, Séverine Begard2, Morvane Colin2, Chloé Lamarre2, Hélène Tran2, Luc Buée2, Joëlle Marie1, Nicolas Sergeant2* & Denis Furling1*
1 Sorbonne Université, Inserm, Institut de myologie, Centre de recherche en myologie, 75013 Paris, France
2 Université de Lille, Inserm, CHU Lille, Lille Neuroscience & Cognition, 59045 Lille, France
3 Sorbonne Paris Cité, Université Paris Descartes, 75005 Paris, France
4 Sorbonne Université, Inserm, Phénotypage du petit animal, 75013 Paris, France
& These authors contributed equally
* Corresponding authors
Reversal of RNA toxicity in myotonic dystrophy via a decoy RNA-binding protein with high affinity for expanded CUG repeats
Ludovic Arandel1,&, Magdalena Matloka1,&, Arnaud F. Klein1, Frédérique Rau1, Alain Sureau1, Michel Ney1, Aurélien Cordier1, Maria Kondili1, Micaela Polay-Espinoza1, Naira Naouar1, Arnaud Ferry1,3, Mégane Lemaitre1,4, Séverine Begard2, Morvane Colin2, Chloé Lamarre2, Hélène Tran2, Luc Buée2, Joëlle Marie1, Nicolas Sergeant2* & Denis Furling1*
1 Sorbonne Université, Inserm, Institut de myologie, Centre de recherche en myologie, 75013 Paris, France
2 Université de Lille, Inserm, CHU Lille, Lille Neuroscience & Cognition, 59045 Lille, France
3 Sorbonne Paris Cité, Université Paris Descartes, 75005 Paris, France
4 Sorbonne Université, Inserm, Phénotypage du petit animal, 75013 Paris, France
& These authors contributed equally
* Corresponding authors
Nature Biomedical Engineering, février 2022
DOI : https://doi.org/10.1038/s41551-021-00838-2