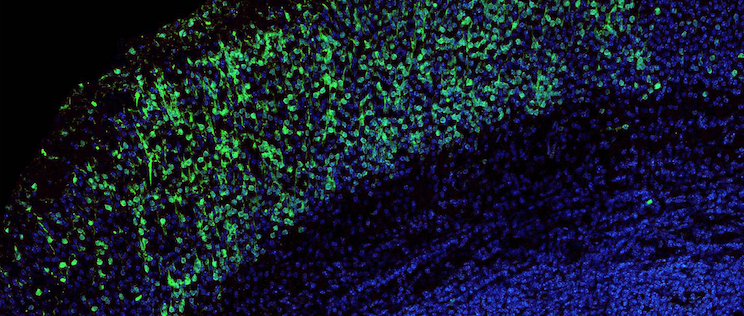
Brain slice showing neurons migrating to the auditory cortex. The nuclei are shown in blue. In green, molecules from the “molecular code” that sends the neurons to this brain region.
©Institut Pasteur
Scientists from the Institut Pasteur, Inserm, the Collège de France and Pierre & Marie Curie University have recently demonstrated that mutations in three genes responsible for Usher syndrome – a hereditary condition that affects both hearing and sight – influence not only the workings of the ear, specifically the function of sensory cells in the cochlea, but also the development of the auditory cortex. Their discovery could explain why some patients, even after being fitted with a cochlear implant (an electro-acoustic device that bypasses the defective cochlea), still have difficulties understanding speech. The findings are reported this week in a paper in the Proceedings of the National Academy of Sciences of the USA.
In most cases of hereditary hearing loss in humans, damage to the auditory sensory organ, the cochlea, is sufficient to account for patients’ hearing impairment. Many forms of hereditary hearing loss affect the hair bundle that acts as the sound receptor for auditory sensory cells. Cochlear implants stimulate the auditory nerve directly, bypassing the need for the sound signal to be processed by the cochlea and restoring a good level of hearing. In some cases, however, patients still have difficulties in understanding speech.
The team led by Prof. Christine Petit[1] from the Genetics & Physiology of Hearing Unit (Institut Pasteur/Inserm/UPMC) – with research by Baptiste Libé-Philippot supervised by group leader Dr. Nicolas Michalski –, working in cooperation with Dr. Christine Métin (Inserm/UPMC), recently identified three genes in mice which, when damaged, affect not only the cochlea but also the auditory cortex, the brain region responsible for analyzing auditory information. These three genes are among the nine currently recognized as causing Usher syndrome (type I and II), the leading hereditary cause of hearing and sight loss. Since damage to the cochlea in these patients prevents the auditory brain from receiving some or all of the acoustic information it normally receives, the cerebral damage had previously gone unnoticed.
The scientists demonstrated that, during embryogenesis, the proteins coded by these three genes are involved in the migration and maturation of some cells destined to become “inhibitory” neurons, which specifically colonize the auditory cortex. These neurons produce parvalbumin and are closely involved in cortical plasticity, which determines the structure and function of neural networks in the cortex based on auditory experience. They also play an important role in the temporal precision of sound detection that is required to understand speech. In mice, a single mutation in one of these three genes prevented this population of neural precursors from entering the developing cortex and reaching the auditory cortex.
The scientists also revealed that these neurons synthesize molecules that act as molecular markers. “These molecules serve as labels that instruct neurons to be sent from their birthplace in the subpallium, in the center of the brain, to the cortical area, their final destination. For the first time, we suggest that inhibitory neuron precursors have a “molecular mailing code”,“ explains Prof. Christine Petit.
These findings therefore demonstrate that hearing loss genes, previously thought to influence just the cochlea, also have another independent role in the development and formation of neural networks in the auditory cortex. In embryogenesis, this role is performed prior to the one played by the same genes in the cochlea.
Prof. Christine Petit’s team is at the forefront of pioneering research into hereditary hearing loss. It previously demonstrated that the proteins coded by the genes that cause type I and type II Usher syndrome are also responsible for the development and workings of the hair bundle, and went on to unravel the related molecular networks. The scientists hope to use these new findings to develop innovative aural rehabilitation methods designed specifically for patients with damage to the auditory cortex.
This research was funded by the French National Research Agency (LIGHT4DEAF [ANR-15-RHUS-0001] and LIFESENSES LabEx [ANR-10-LABX-65]), the European Commission (ERC-2011-ADG_294570), the BNP Paribas Foundation, the FAUN Stiftung (Suchert Foundation) and the LHW-Stiftung (CP), and the “scientific discovery” award and SEIZEAR grant from the “Agir Pour l’Audition” foundation (NM).
[1] Prof. Christine Petit is a Professor at the Collège de France and at the Institut Pasteur.
These contents could be interesting :


Medias
Sources
Auditory cortex interneuron development requires cadherins operating hair-cell mechanoelectrical transduction Baptiste Libé-Philippot (1,2,3), Vincent Michel (1,2,3), Jacques Boutet de Monvel (1,2,3), Sébastien Le Gal (1,2,3), Typhaine Dupont (1,2,3), Paul Avan (4,5,6), Christine Métin (7,8,9)*, Nicolas Michalski (1,2,3)* and Christine Petit (1,2,3,10,11)*.
- Unité de Génétique et Physiologie de l’Audition, Institut Pasteur, 75015 Paris, France
- UMRS 1120, Inserm, 75015 Paris, France
- Sorbonne Universités, UPMC Université Paris 06, Complexité du Vivant, 75005 Paris, France
- Laboratoire de Biophysique Sensorielle, Université d’Auvergne, 63000 Clermont-Ferrand, France
- UMR 1107,Inserm, 63000 Clermont-Ferrand, France
- Centre Jean Perrin, 63000 Clermont-Ferrand, France
- Institut du Fer à Moulin, 75005 Paris, France
- UMRS 839, Inserm, 75005 Paris, France
- Sorbonne Universités, UPMC Université Paris 06, Cerveau-Cognition-Comportement (ED3C), 75005 Paris, France
- Syndrome de Usher et Autres Atteintes Rétino-Cochléaires, Institut de la Vision, 75012 Paris, France
- Collège de France, 75005 Paris, France