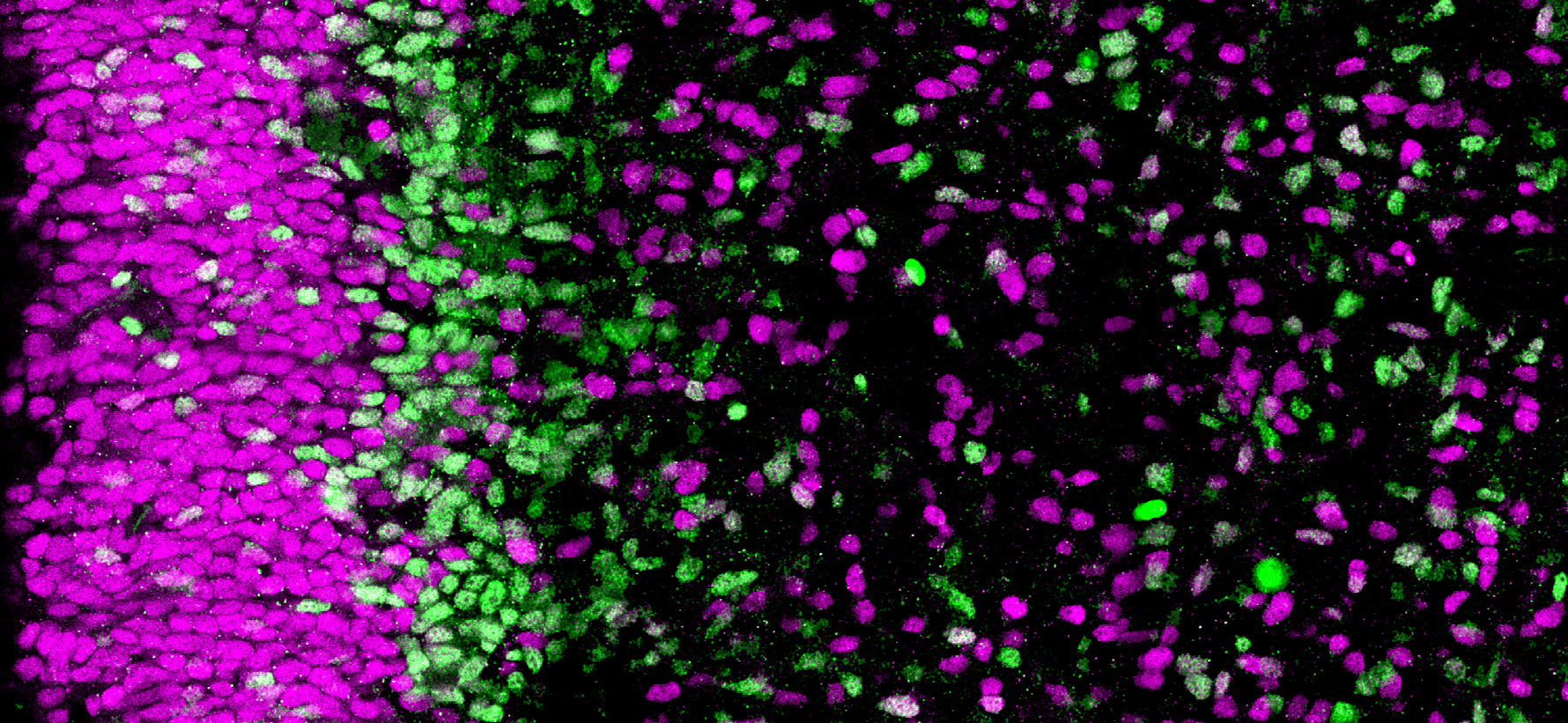
Section of the human brain (cortex). The progenitor cells in magenta are less engaged in neuronal differentiation than those in green © Monia Barnat/Grenoble Institut des Neurosciences/Inserm, Université Grenoble Alpes
Huntington’s disease is a genetic neurological condition that usually manifests in adulthood. Teams of researchers and clinicians from Inserm, Université Grenoble Alpes, Sorbonne Université, CNRS and AP-HP at the Grenoble Institute of Neuroscience and the Brain Institute, have discovered abnormalities in the brains of human embryos that carry the mutation responsible for Huntington’s disease. This research, to be published in Science, looks at the mechanisms of the silent progression of the disease and when and how to treat patients in the future.
Huntington’s is a rare and hereditary genetic disease of the central nervous system, which usually manifests between 30 and 50 years of age in the form of gradually-worsening psychiatric, cognitive and motor disorders. It is caused by a mutation of the gene coding for the huntingtin protein and is transmitted in an “autosomal dominant” manner: inheriting just one copy of the defective gene is enough to develop the condition. Around 18,000 people in France are concerned, of whom 6,000 are already symptomatic and around 12,000 possess the gene carrying the mutation but are asymptomatic.
The teams of Sandrine Humbert, Inserm research director at the Grenoble Institute of Neuroscience (Inserm/Université Grenoble Alpes), and Alexandra Durr, university professor-hospital practitioner at Sorbonne Université, Pitié Salpêtrière hospital – AP-HP, and the Brain Institute (Inserm/Sorbonne Université/CNRS/AP-HP), are interested in the early stages of Huntington’s disease and the long period that precedes the onset of symptoms. In new research published in Science, they looked at when brain abnormalities could develop.
The teams studied the brains of 13-week-old human embryos donated by parents following medical termination of pregnancy. They observed several differences between the embryos that carry the mutation of the gene coding for huntingtin and those that do not.
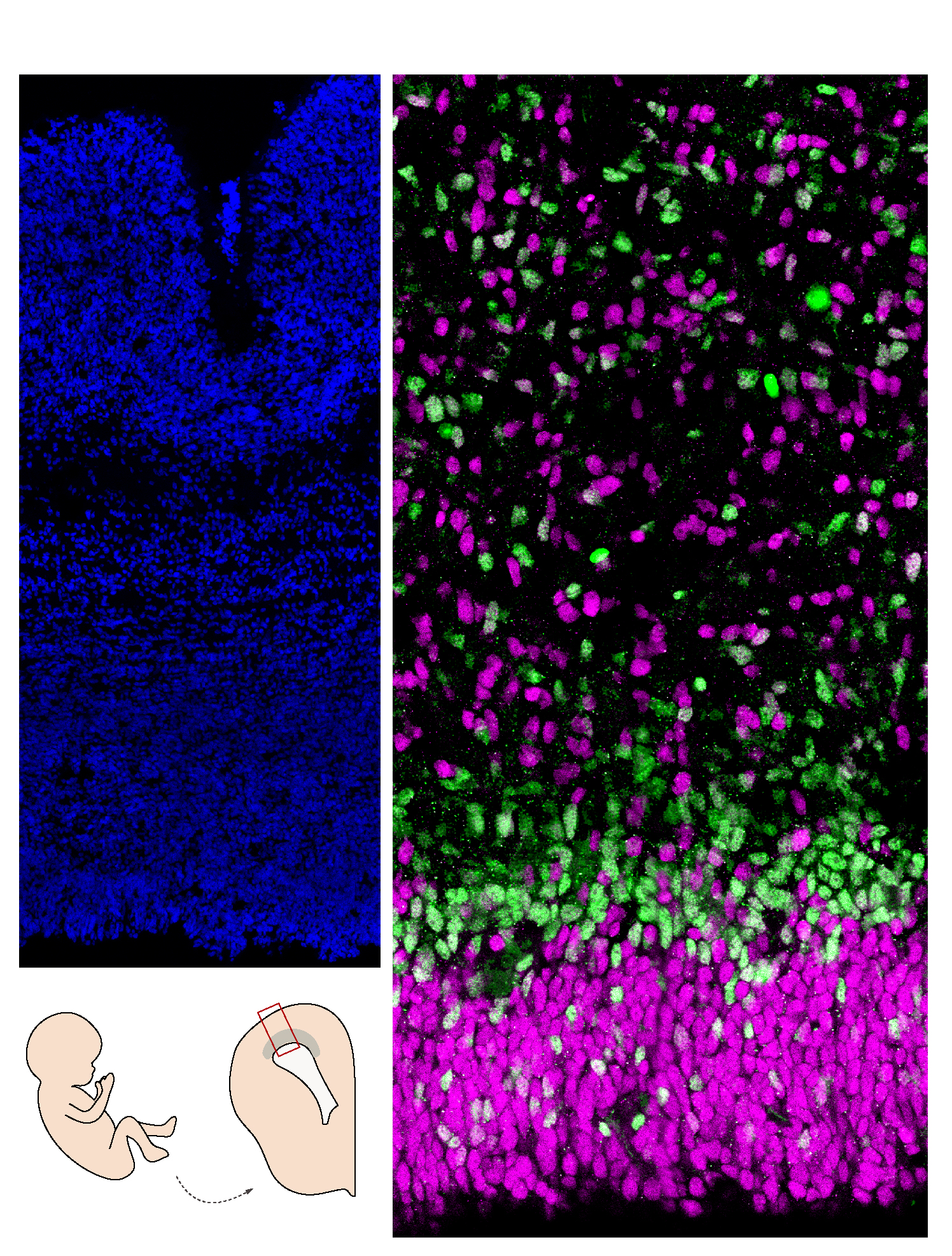
In the carriers, the pathological huntingtin protein is abnormally localized in the progenitor cells that generate the neurons of the cortex. This mislocalization is linked, among other things, to problems of junction protein localization in these cells and to alterations in the size, density and orientation of the
cilium, an organelle essential to the functioning of these cells. These abnormalities disrupt the “division-differentiation” balance of the progenitor cells, which are derived from a reservoir of dividing cells, some of which differentiate into neurons while the others continue to divide in order to provide new progenitor cells. In the embryos that carry the mutation, these progenitor cells start differentiating more quickly at the expense of the reservoir of dividing cells.
The researchers repeated the experiment with a mouse model of Huntington’s disease at an equivalent stage of embryonic development and found the same abnormalities. This meant they could validate this animal model in order to further explore the early mechanisms of the disease at other stages of embryonic development or after birth.
“This is the first time that abnormalities of brain development have been identified in this disease. Abnormalities which are also relatively large and extensive, although we are not yet able to determine their direct consequences,” clarify Humbert and Durr, who led this research.
But why do the carriers of the mutation show no symptoms until later down the line? “At this stage, our hypothesis is that very early on the brain implements compensatory mechanisms that allow normal functioning. It could be the same in people who carry mutations linked to other types of degeneration, such as Alzheimer’s disease or amyotrophic lateral sclerosis,” specify the researchers.
They will now continue to describe brain development in mouse models of Huntington’s disease, try to understand how these early defects contribute to the adult disease, and how their compensation could be regulated during the silent symptomless period. “This discovery also has important implications for how and when disease-modifying therapies should from now on be considered,” they conclude.

Human brain section (cortex). On the left, the nuclei are marked in blue; on the right, the progenitor cells in magenta are less engaged in neuronal differentiation than those in green.
©Monia Barnat/Grenoble Institut des Neurosciences/Inserm, Université Grenoble Alpes
These contents could be interesting :