Researcher Contact
Chercheur Inserm
Jocelyn Laporte
Institut de génétique et de biologie moléculaire et cellulaire (IGBMC)
Equipe "Physiopathologie des maladies neuromusculaires"
Tel : 03 88 65 34 15
Inserm and CNRS researchers from the Institute of Genetics and Molecular and Cellular Biology (Inserm/CNRS/Université de Strasbourg) have discovered how myotubularin – a protein deficient in myotubular myopathy – interacts with amphiphysin 2 and suggest targeting the latter in order to treat patients. This research was published on March 20, 2019 in Science Translational Medicine.
When exploring the interactions of myotubularin (coded by the MTM1 gene) with another protein, amphiphysin 2 (coded by the BIN1 gene), which is also expressed in the muscles and involved in similar myopathies, Inserm’s “Pathophysiology of neuromuscular diseases” team, in conjunction with the CNRS at the Institute of Genetics and Molecular and Cellular Biology (CNRS/Inserm/Université de Strasbourg), discovered how these proteins work together and suggests a new therapeutic target. Previous research had shown that myotubularin and amphiphysin 2 can physically interact by binding to each other.
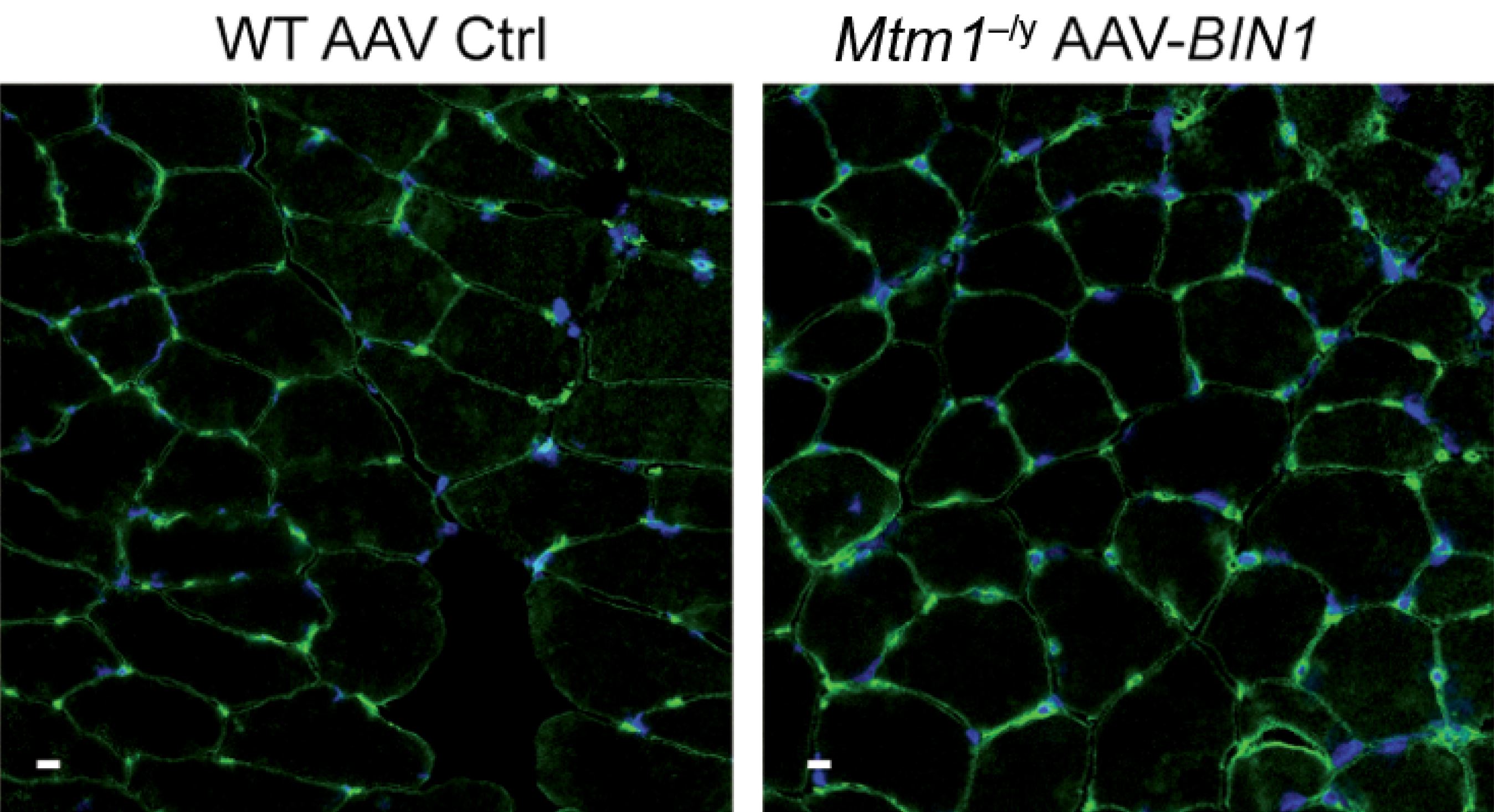
To explore this functional link between the two, the researchers developed a model of MTM1-deficient transgenic mice and crossed these animals with other mice – some of which do not express BIN1 and some of which, on the contrary, overexpress it. They were unable to obtain any animals deficient in both MTM1 and BIN1, proving that at least one of the two proteins is necessary for muscle-fiber development and fetal survival. Conversely – and this came as a pleasant surprise – the overexpression of BIN1 made it possible to correct the myopathy linked to the MTM1 deficiency and obtain life expectancy equivalent to that of wild animals. Upon closer analysis of the muscles, the researchers observed satisfactory muscle-fiber organization and size with good cell adhesion, thereby leading to the hypothesis that MTM1 is an in vivo activator of the bin1 protein and that large quantities of the latter could make it possible to “do without” MTM1.
To verify whether BIN1 is a good therapeutic target, the researchers went on to conduct a gene therapy experiment in MTM1-deficient mice. They administered the human BIN1 gene using an AAV viral vector by systemic (intraperitoneal) injection following the birth of the rodents. A procedure that markedly reduced the symptoms of the condition and increased the survival of the diseased mice to that of healthy mice.
Chercheur Inserm
Jocelyn Laporte
Institut de génétique et de biologie moléculaire et cellulaire (IGBMC)
Equipe "Physiopathologie des maladies neuromusculaires"
Tel : 03 88 65 34 15
Amphiphysin 2 (BIN1) modulation rescues MTM1 myotubular myopathy and prevents focal adhesion defects
Valentina M. Lionello1,2,3,4, Anne-Sophie Nicot1,2,3,4,8,9, Maxime Sartori1,2,3,4, Christine Kretz1,2,3,4, Pascal Kessler1,2,3,4, Suzie Buono1,2,3,4, Sarah Djerroud1,2,3,4, Nadia Messaddeq1,2,3,4, Pascale Koebel1,2,3,4, Ivana Prokic1,2,3,4,, Yann Hérault1,2,3,4, Norma B Romero5,6,7, Jocelyn Laporte1,2,3,4,*,†, Belinda S. Cowling1,2,3,4,†
1Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC), Illkirch, France
2INSERM U1258, Illkirch, France
3CNRS UMR7104, Illkirch, France
4Strasbourg University, Illkirch, France
5Université Sorbonne, UPMC Univ Paris 06, INSERM UMRS974, CNRS FRE3617, Center for
Research in Myology, GH Pitié-Salpêtrière, 47 Boulevard de l’hôpital, 75013 Paris, France
6Centre de référence de Pathologie Neuromusculaire Paris-Est, Institut de Myologie, GHU Pitié-
Salpêtrière, Assistance Publique-Hôpitaux de Paris, Paris, France
7Neuromuscular Morphology Unit, Institut de Myologie, GHU Pitié-Salpêtrière, Assistance
Publique-Hôpitaux de Paris, Paris, France
8Grenoble Institute des Neurosciences, Université Grenoble Alpes, Grenoble, France
9INSERM, U1216, Grenoble, France
Science Translational Medicine, 20 Mar 2019
http://dx.doi.org/10.1126/scitranslmed.aav1866