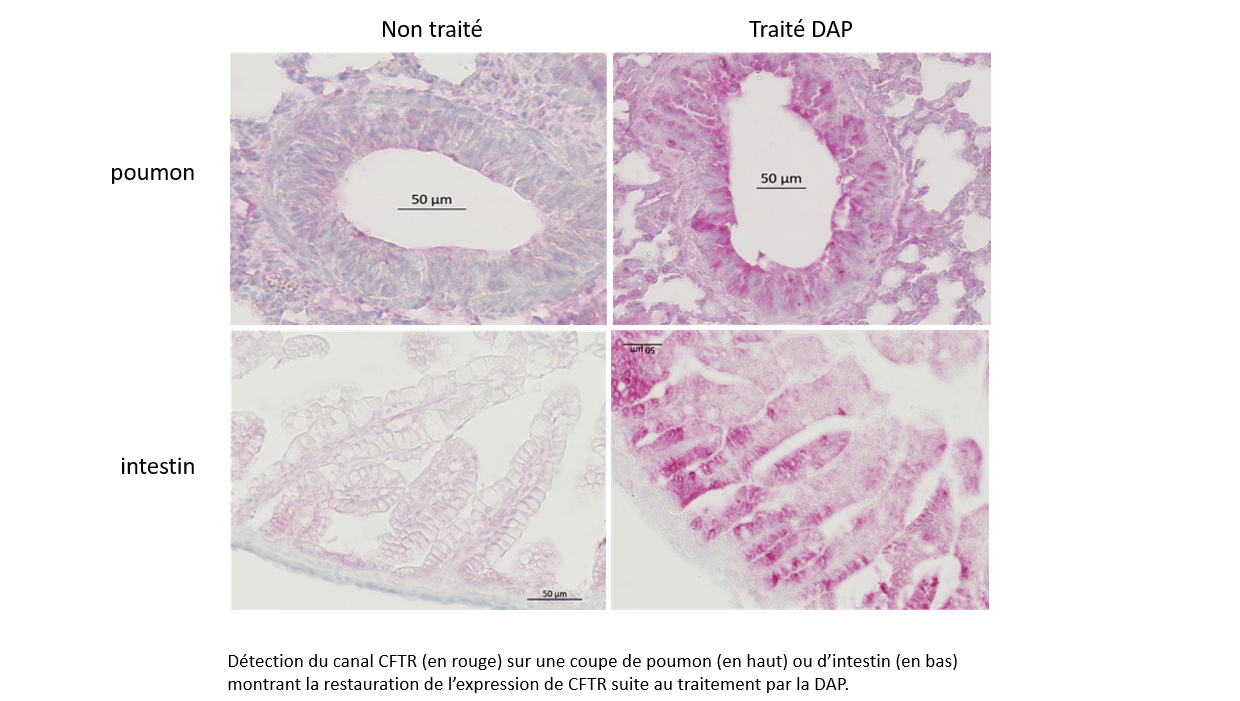
Use of 2,6-diaminopurine as a potent suppressor of UGA premature stop codons in cystic fibrosis
Catherine Leroy1,2,ꝉ, Sacha Spelier3,ꝉ, Nadège Charlene Essonghe4,5, Virginie Poix1,2, Rebekah Kong1,2, Patrick Gizzi6, Claire Bourban6, Séverine Amand7, Christine Bailly7, Romain Guilbert8, David Hannebique8, Philippe Persoons8, Gwenaëlle Arhant1,2, Anne Prévotat9, Philippe Reix10, Dominique Hubert11, Michèle Gérardin12, Mathias Chamaillard4, Natalia Prevarskaya4,5, Sylvie Rebuffat7, George Shapovalov4,5, Jeffrey Beekman3 and Fabrice Lejeune1,2*
1 Univ. Lille, CNRS, Inserm, UMR9020-U1277 – CANTHER – Cancer Heterogeneity Plasticity and Resistance to Therapies, F-59000 Lille, France
2 Unité tumorigenèse et résistance aux traitements, Institut Pasteur de Lille, F-59000 Lille, France
3Pediatric Respiratory Medicine, Wilhelmina Children’s Hospital, University Medical Center, Utrecht University, 3584 EA Utrecht, The Netherlands; Regenerative Medicine Utrecht, University Medical Center, Utrecht University, 3584 CT Utrecht, The Netherlands; Centre for Living Technologies, University Medical Center, Utrecht University, 3584 CT Utrecht, The Netherlands
4Univ. Lille, Inserm, U1003 – PHYCEL – Physiologie Cellulaire, F-59000 Lille, France
5Laboratory of Excellence, Ion Channels Science and Therapeutics, 59655 Villeneuve d’Ascq, France
6Plateforme de chimie biologique intégrative de Strasbourg, UAR 3286 CNRS – Université de Strasbourg, Illkirch, France
7Muséum national d’Histoire naturelle, Centre national de la Recherche scientifique, Laboratory Molecules of Communication and Adaptation of Microorganisms (MCAM), UMR 7245 CNRS-MNHN, CP 54, 57 rue Cuvier, 75005 Paris, France.8Institut Pasteur de Lille – PLEHTA (Plateforme d’expérimentation et de Haute Technologie Animale), 59019 Lille, France.
9 Univ. Lille, Clinique des Maladies Respiratoires, CRCM Hôpital Calmette, CHRU Lille, France.
10 CRCM pédiatrique Lyon. Hôpital Femme Mère Enfant. Hospices Civils de Lyon. UMR 5558 (EMET). CNRS, LBBE, Université de Lyon, Villeurbanne France.
11Pulmonary Department and Adult CF Centre, Cochin Hospital, AP-HP, Paris, France.
12 CF pediatric centre, Robert Debré hospital, AP-HP, Paris, France.
Molecular Therapy, janvier 2023
DOI : https://doi.org/10.1016/j.ymthe.2023.01.014