
Contact scientifique
Chercheur Inserm
Jocelyn Laporte
Institut de génétique et de biologie moléculaire et cellulaire (IGBMC)
Equipe "Physiopathologie des maladies neuromusculaires"
Tel : 03 88 65 34 15
Des chercheurs Inserm et CNRS de l’Institut de génétique et de biologie moléculaire et cellulaire (Inserm/CNRS/Université de Strasbourg) ont découvert comment la myotubularine, protéine déficitaire dans la myopathie myotubulaire, interagit avec l’amphiphysine 2 et proposent de cibler cette dernière pour traiter les patients. Ces travaux sont parus le 20 mars 2019 dans Science Translational Medicine.
En explorant les interactions de la myotubularine, protéine codée par le gène MTM1, avec une autre protéine, l’amphiphysine 2 codée par le gène BIN1, également exprimée dans les muscles et impliquée dans des myopathies similaires, l’ équipe Inserm « Physiopathologie des maladies neuromusculaires », avec la collaboration du CNRS, à l’Institut de génétique et de biologie moléculaire et cellulaire (CNRS/Inserm/Université de Strasbourg) a découvert comment ces protéines travaillent ensemble et propose une nouvelle cible thérapeutique. De précédents travaux avaient en effet montré que la myotubularine et l’amphiphysine 2 peuvent interagir physiquement en se liant l’une à l’autre.
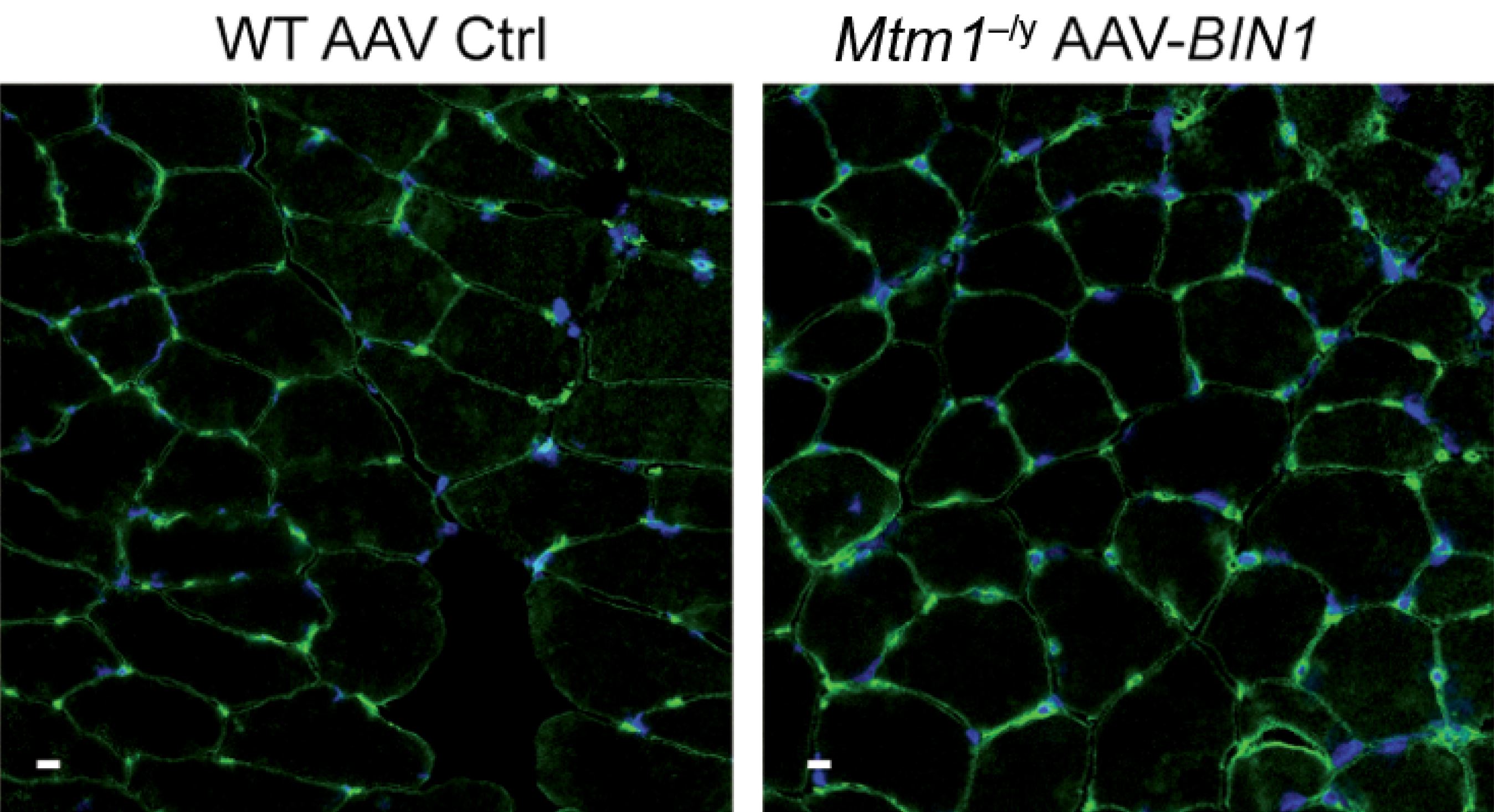
Pour explorer le lien fonctionnel entre les deux, les chercheurs ont développé un modèle de souris transgéniques déficitaires en MTM1 et ont croisé ces animaux avec d’autres souris dont certaines n’expriment pas BIN1 et d’autres qui au contraire, surexpriment ce gène. Ils n’ont obtenu aucun animal déficitaire à la fois en MTM1 et BIN1, prouvant qu’au moins l’une des deux protéines est nécessaire au développement des fibres musculaires et à la survie du fœtus. A l’inverse, et c’est la bonne surprise, la surexpression de BIN1 a permis de corriger la myopathie liée au déficit de MTM1 et d’obtenir une espérance de vie équivalente aux animaux sauvages. En analysant de plus près les muscles, les chercheurs ont constaté une organisation et une taille correcte des fibres musculaires avec une bonne adhésion des cellules entre elles. Ils ont donc fait l’hypothèse que MTM1 est un activateur de la protéine bin1 in vivo, et que fournir cette dernière en grande quantité pourrait permettre de se « passer » de MTM1.
Pour vérifier si BIN1 est une bonne cible thérapeutique, ils ont mené dans un second temps une expérience de thérapie génique chez des souris déficitaires en MTM1. Ils ont administré le gène BIN1 humain grâce à un vecteur viral de type AAV par injection systémique (intra-péritonéale) après la naissance des rongeurs. Cette intervention a nettement réduit les symptômes de la maladie et prolongé la survie des souris malades, à hauteur de celle de souris saines.
Chercheur Inserm
Jocelyn Laporte
Institut de génétique et de biologie moléculaire et cellulaire (IGBMC)
Equipe "Physiopathologie des maladies neuromusculaires"
Tel : 03 88 65 34 15
Amphiphysin 2 (BIN1) modulation rescues MTM1 myotubular myopathy and prevents focal adhesion defects
Valentina M. Lionello1,2,3,4, Anne-Sophie Nicot1,2,3,4,8,9, Maxime Sartori1,2,3,4, Christine Kretz1,2,3,4, Pascal Kessler1,2,3,4, Suzie Buono1,2,3,4, Sarah Djerroud1,2,3,4, Nadia Messaddeq1,2,3,4, Pascale Koebel1,2,3,4, Ivana Prokic1,2,3,4,, Yann Hérault1,2,3,4, Norma B Romero5,6,7, Jocelyn Laporte1,2,3,4,*,†, Belinda S. Cowling1,2,3,4,†
1Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC), Illkirch, France
2INSERM U1258, Illkirch, France
3CNRS UMR7104, Illkirch, France
4Strasbourg University, Illkirch, France
5Université Sorbonne, UPMC Univ Paris 06, INSERM UMRS974, CNRS FRE3617, Center for
Research in Myology, GH Pitié-Salpêtrière, 47 Boulevard de l’hôpital, 75013 Paris, France
6Centre de référence de Pathologie Neuromusculaire Paris-Est, Institut de Myologie, GHU Pitié-
Salpêtrière, Assistance Publique-Hôpitaux de Paris, Paris, France
7Neuromuscular Morphology Unit, Institut de Myologie, GHU Pitié-Salpêtrière, Assistance
Publique-Hôpitaux de Paris, Paris, France
8Grenoble Institute des Neurosciences, Université Grenoble Alpes, Grenoble, France
9INSERM, U1216, Grenoble, France
Science Translational Medicine, 20 Mar 2019
https://dx.doi.org/10.1126/scitranslmed.aav1866