La morphine est utilisée depuis plusieurs siècles pour soulager les douleurs intenses. Ses propriétés antidouleur sont toutefois accompagnées d’effets secondaires importants. La morphine mime l’action de molécules produites naturellement par le cerveau (les endorphines). Pourquoi alors a-t-elle des effets secondaires aussi délétères ? L’explication vient d’être apportée par l’équipe de Sébastien Granier, chercheur à l’Institut de génomique fonctionnelle (Inserm/CNRS/Universités de Montpellier 1et 2) et ses collaborateurs américains. La structure 3D des récepteurs du cerveau sur lesquels se fixent la morphine ou les endorphines, est probablement différente selon que l’une ou l’autre des molécules s’y fixe. La réponse de l’organisme, va, de fait, être totalement modifiée. Grâce à cette découverte, les chercheurs espèrent réussir à conserver les effets bénéfiques de la morphine sans pour autant induire d’effets secondaires.
Ces travaux réalisés sont publiés dans la revue Nature datée du 21 mars 2012
L’opium, produit naturel extrait du pavot (Papaver somniferum), est une des plus anciennes drogues connues par l’Homme pour ses propriétés psychotropes, sédatives et analgésiques. Ces effets sont induits par son composant majeur, la morphine, qui est largement utilisée de nos jours en clinique pour soulager la douleur.
L’action de la morphine est relayée par les récepteurs µ-opiacés exprimés à la surface des cellules du système nerveux central. Ces récepteurs font partie d’une superfamille de protéines, les récepteurs couplés aux protéines G (RCPG) qui sont la cible d’environ 30 % des médicaments actuellement sur le marché.
Au niveau moléculaire, la morphine, en se liant aux récepteurs µ-opiacés, mime l’action de molécules produites naturellement dans le cerveau : les endorphines. Cependant son utilisation en clinique est limitée par deux effets. D’une part, le développement d’un phénomène de tolérance oblige à augmenter la dose de morphine au fur et à mesure des injections répétées pour obtenir le même effet thérapeutique. D’autre part, la consommation de morphine entraine un phénomène de dépendance comme la drogue (l’héroïne, forme acétylée de la morphine, étant l’exemple le plus édifiant). De plus, la morphine provoque de graves effets secondaires : dépression respiratoire, constipation, dépendance physique et psychique. Ces effets délétères s’expliquent notamment par le fait que la morphine déclenche une réponse cellulaire différente de celle induite par les endorphines. La morphine et les endorphines se liant au même récepteur, ces deux molécules stabiliseraient les récepteurs µ-opiacés dans des conformations spatiales distinctes à l’origine des différences de réponses biologiques.
« Dans le but de développer des molécules conservant les effets bénéfiques de la morphine sans pour autant induire d’effets secondaires, il est donc indispensable de comprendre les bases structurales de l’action de la morphine et des opiacés en général » explique Sébastien Granier, chercheur à l’Inserm et principal auteur de ce travail.
Le travail réalisé par Sébastien Granier et l’équipe de Brian Kobilka à Stanford, a permis de résoudre la structure tridimensionnelle du récepteur µ-opiacé lorsqu’il est associé à une molécule présentant une structure chimique proche de celle de la morphine.

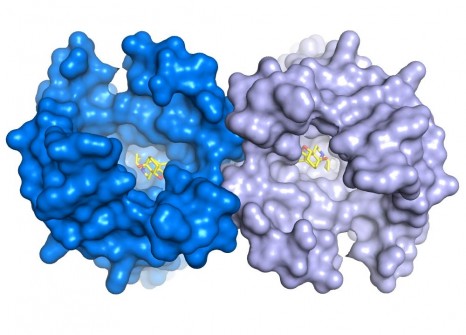
La structure 3D ainsi visualisée montre que la zone spécifique du récepteur où se lie la molécule opioïde est largement ouverte vers le milieu extérieur, ce qui explique la rapidité d’action de ces composés.
Cette structure révèle aussi une caractéristique très importante dans le fonctionnement de ce récepteur : la formation d’un dimère de récepteur (1). « C’est la toute première fois que nous réussissons à visualiser la structure 3D d’un tel complexe pour cette famille de récepteur » ajoute le chercheur. Cette structure d’un dimère de récepteurs µ-opiacés ouvre de nouvelles pistes pour étudier ce phénomène et mieux comprendre ces implications fonctionnelles.
La résolution de la structure 3D du récepteur µ-opiacé, clé du traitement de la douleur et des addictions, pourrait à terme mener à la conception de nouveaux médicaments analgésiques dépourvues d’effets secondaires.
Note
(1) Molécule issue de “l’association” de deux molécules identiques