©Adobe Stock
Un véritable capteur : une équipe de chercheurs de l’Inserm et du CNRS au sein de l’Institut de pharmacologie et de biologie structurale (IPBS, CNRS/Université Toulouse III – Paul Sabatier) a identifié une protéine capable de détecter divers allergènes dans les voies respiratoires à l’origine de crises d’asthme. Cette étude co-dirigée par Corinne Cayrol et Jean-Philippe Girard est publiée dans la revue Nature Immunology le 19 mars 2018. Elle augure des avancées dans le traitement des maladies allergiques.
Quel est le point commun entre moisissures, pollens et cafards ? Bien qu’ils appartiennent à trois règnes distincts du monde vivant, ils peuvent déclencher des crises d’asthme chez les personnes sensibles. En dépit de compositions très différentes, ils partagent un point commun : ils contiennent tous des enzymes appelées protéases.
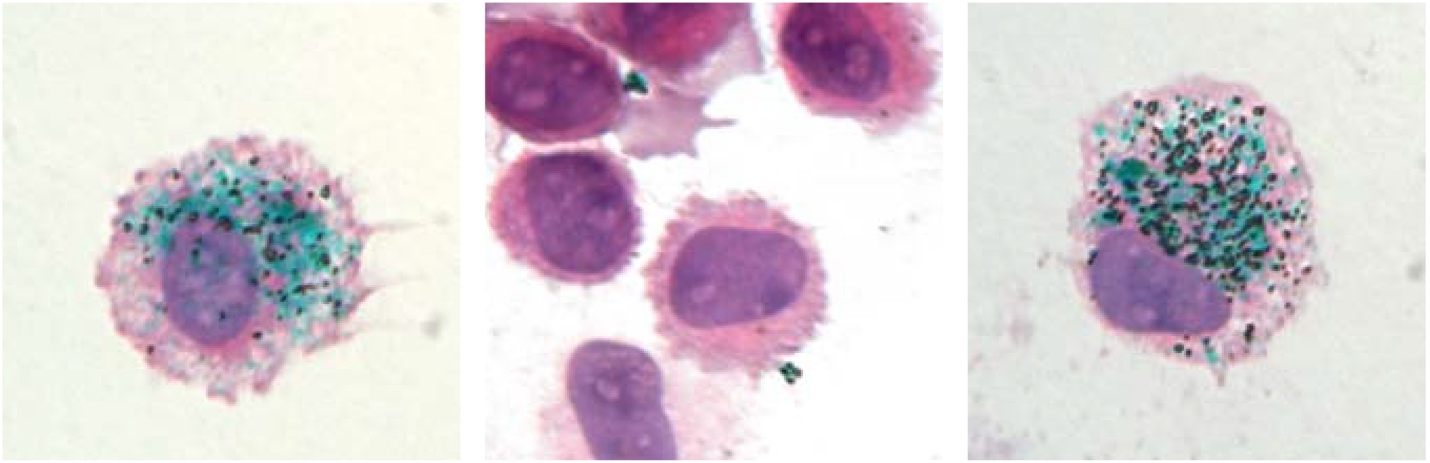
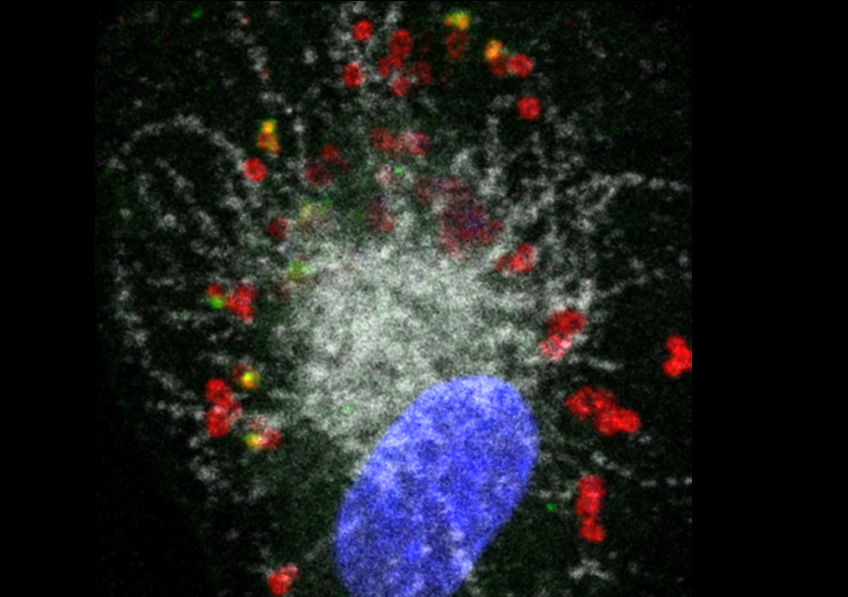
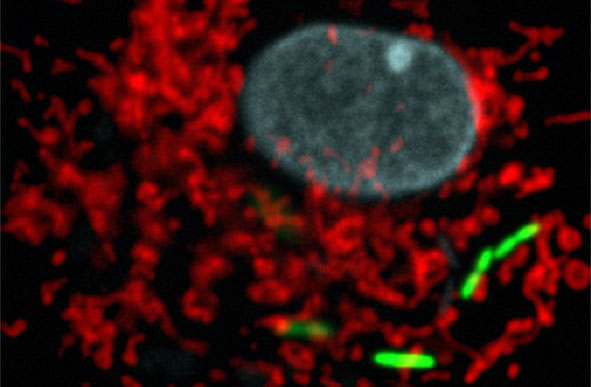
L’équipe de l’IPBS vient d’identifier une protéine humaine réagissant à bon nombre d’allergènes de l’environnement : l’interleukine-33 (IL-33). Lorsqu’ils arrivent dans les voies respiratoires, les allergènes libèrent leurs protéases qui découpent l’IL-33 en fragments hyperactifs à l’origine des réactions en chaîne responsables des symptômes allergiques.
Or, il s’agirait d’un mécanisme général de déclenchement des réactions allergiques. En effet, l’IL-33 s’est montrée capable de détecter chacun des 14 allergènes testés, parmi lesquels certains sont présents dans l’air ambiant (plusieurs types de pollens, des acariens, des spores de champignons) et d’autres impliqués dans l’asthme professionnel (comme la subtilisine utilisée dans des détergents).
Ces résultats sont d’autant plus importants qu’ils établissent un lien direct entre génétique et environnement. En effet, le gène codant pour l’IL-33 est reconnu comme étant l’un des principaux gènes de prédisposition à l’asthme chez l’humain.
Des essais cliniques en cours ont d’ailleurs pris pour cible cette protéine. Une stratégie que vient confirmer cette découverte d’un mécanisme unique de détection par l’IL-33 des allergènes aériens. Empêcher la production des fragments hyperactifs de l’IL-33 après une exposition aux allergènes pourrait, par exemple, permettre de limiter les réactions allergiques sévères chez les patients asthmatiques.
©Corinne Cayrol et Jean-Philippe Girard / IPBS / CNRS-Université Toulouse III – Paul Sabatier
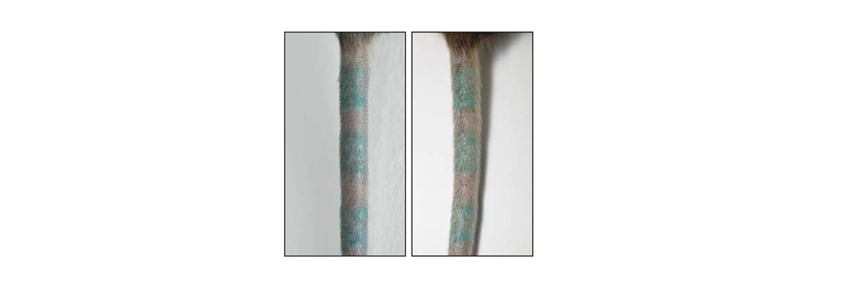
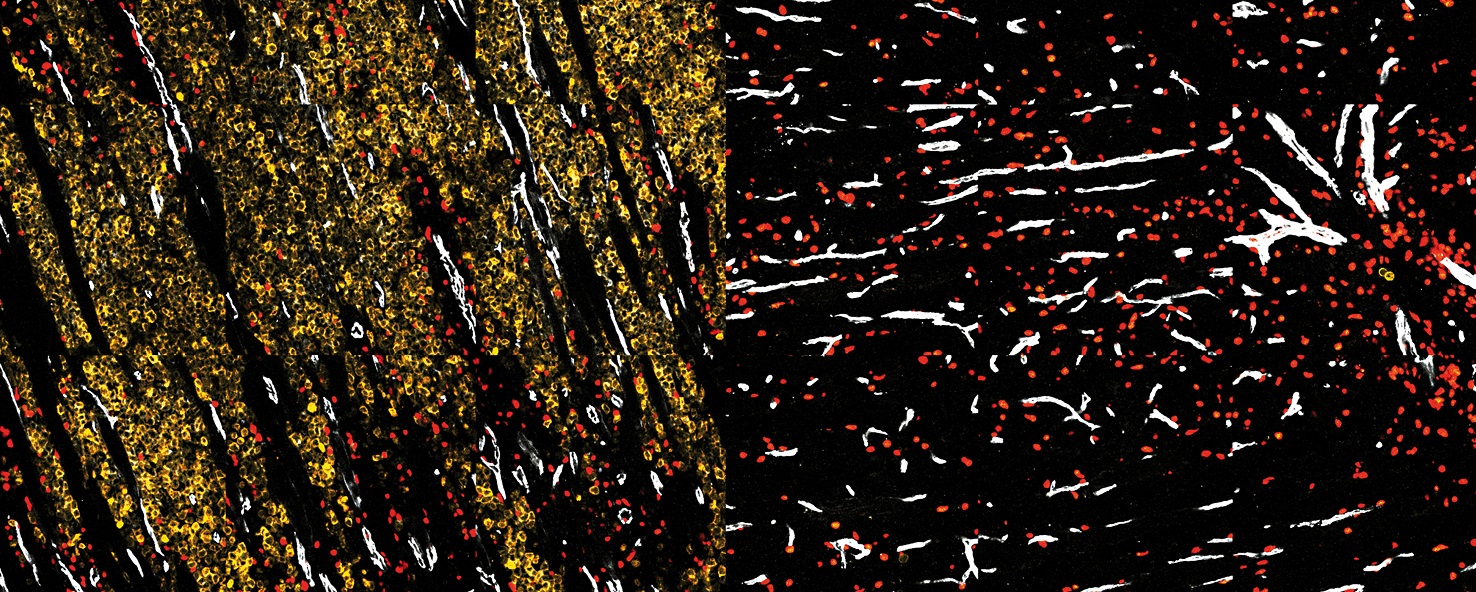
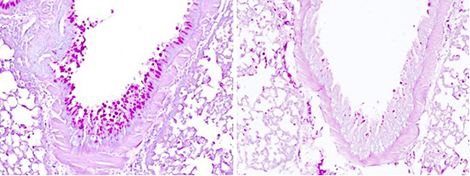
Production de mucus dans le poumon après inhalation d’un allergène (coupes de poumon, coloration du mucus en rose magenta).
L’hyperproduction de mucus est l’une des caractéristiques de l’asthme allergique. La protéine IL-33, un facteur majeur de prédisposition à l’asthme chez l’homme, détecte l’activité protéase de l’allergène. Elle s’en trouve activée et déclenche une cascade de réactions, dont la production de mucus, associées à l’asthme et aux autres maladies allergiques. Lorsque l’activation de l’IL-33 est bloquée (à droite), la réaction n’est pas déclenchée.