Les enfants prématurés présentent souvent des troubles digestifs en raison de l’immaturité de leur système digestif, en particulier l’intestin. En étudiant la maturation du système nerveux entérique, acteur clé dans le contrôle des fonctions intestinales, Michel Neunlist, et son équipe de l’unité Inserm 913 « Neuropathies du système nerveux entérique et pathologies digestives : implication des cellules gliales entériques » de l’Institut des Maladies de l’Appareil Digestif de Nantes, ont mis au jour une importante cible thérapeutique : les neurones cholinergiques, qui libèrent l’acétylcholine. Leurs travaux paraissent dans l’édition d’août de l’American Journal of Physiology.
Jusqu’au terme de la grossesse, et dans les mois qui suivent la naissance, différents organes chez l’enfant poursuivent leur maturation et leur développement. C’est le cas de l’intestin dont la motricité et l’activité de la barrière intestinale (perméabilité, immunité, …) ne deviennent totalement fonctionnelles qu’au cours de la période postnatale, moment clé de la vie. Pendant les premiers mois, les enfants nés prématurément ont un intestin très immature et présentent des troubles fonctionnels digestifs, tels que des ralentissements du transit et des ballonnements. En conséquence, leur alimentation est difficile et ils sont, ainsi, enclins à développer des pathologies plus graves. Une meilleure compréhension des mécanismes de maturation du système digestif est indispensable pour tenter de guérir les troubles rencontrés chez ces enfants.
Le système nerveux entérique : un élément clé dans le contrôle des fonctions intestinales
Dans cette étude, Michel Neunlist et son équipe ont concentré leurs recherches sur un des éléments phares du contrôle des fonctions intestinales : le système nerveux entérique (SNE). Le SNE, dont la réelle implication et le développement dans la période post natale restent peu connus, est constitué du plexus muqueux et du plexus myentérique. Ce dernier contrôle plus particulièrement la motricité intestinale en libérant des neuromédiateurs (acétylcholine, monoxyde d’azote…). On parle du phénomène de transmission neuromusculaire. Ces substances chimiques délivrées par les neurones vont induire une activité coordonnée du muscle intestinal, conduisant à la propagation du bol alimentaire.
Les chercheurs ont mené leurs travaux sur le côlon du raton nouveau-né, très bon modèle immature dans l’étude des dysfonctionnements gastrointestinaux chez les enfants prématurés. Lors de la période postnatale, ils ont observé des changements dans la morphologie des neurones du plexus myentérique et, plus particulièrement, une augmentation de la proportion de neurones cholinergiques, synthétisant et libérant l’acétylcholine. Cette évolution du nombre de ces neurones est associée au développement de la transmission neuromusculaire et à la mise en place d’une activité motrice du côlon, in vivo et ex vivo.
« Notre étude a permis d’identifier les circuits cholinergiques du système nerveux entérique, comme un nouvel élément clef nécessaire à la mise en place de la motricité intestinale post-natale. Cette population neuronale apparaît comme une nouvelle cible thérapeutique dans la prise en charge des troubles de la motricité intestinale chez le nouveau-né », précise Michel Neunlist.
Ce laboratoire avait récemment démontré que le butyrate, produit par la fermentation bactérienne de fibres alimentaires, accélérait le transit chez l’adulte en augmentant la proportion de neurones cholinergiques. Ces travaux fondent donc les bases de l’utilisation d’approches thérapeutiques utilisant des facteurs nutritionnels ou leurs dérivés tels que le butyrate.

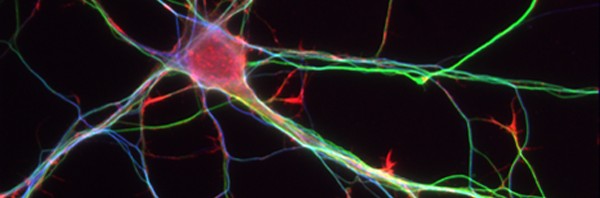
Au cours des premières semaines de vie du raton, l’évolution de la densité des neurones du plexus myentérique (vert) et l’augmentation de la proportion de neurones cholinergiques (rouge) sont corrélées au développement d’une activité motrice dans le colon. © Unité Inserm 913