La peur est une réponse adaptative essentielle à la survie de nombreuses espèces. Cette adaptation comportementale peut être innée ou bien être la conséquence d’un apprentissage au cours duquel un animal apprend qu’un stimulus prédit un évènement désagréable. De nombreuses données indiquent que l’amygdale, une structure particulière du cerveau, est fortement impliquée au cours de l’apprentissage de la peur dite « apprise ». Cependant les circuits neuronaux sous jacents restaient encore largement inconnus jusqu’à présent.
Aujourd’hui, les travaux associant plusieurs équipes suisses, allemandes et un chercheur de l’Unité Inserm 862 à Bordeaux « Neurocentre Magendie« , ont permis d’identifier pour la première fois des circuits neuronaux distincts au sein du noyau central de l’amygdale, spécifiquement impliqués dans l’acquisition et le contrôle des réponses comportementales de peur. Le détail de ces résultats est publié dans la revue Nature, datée de cette semaine.
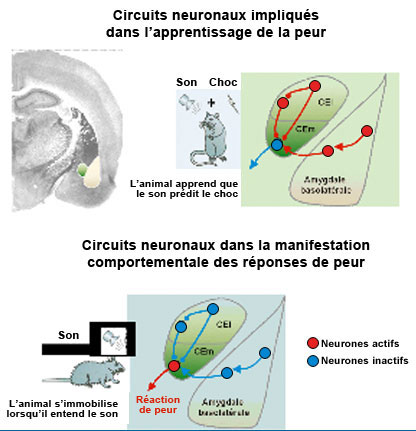
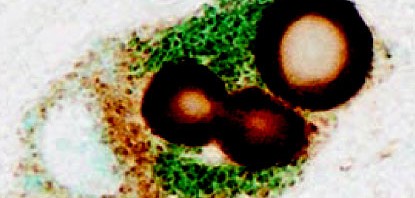
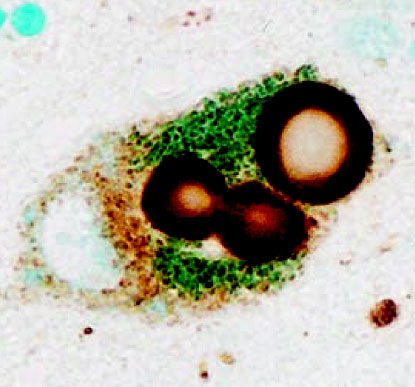
Identification des nouveaux circuits neuronaux inhibiteurs situés au sein du noyau central latéral (CEl) et médian (CEm) de l’amygdale impliqués dans l’apprentissage et la manifestation comportementale des réponses de peur © C. Herry/Inserm
Dans cette étude, des souris de laboratoires ont tout d’abord été soumises à une tâche comportementale simple qui consiste à apprendre qu’un stimulus sonore prédit l’arrivée d’un évènement désagréable. A la suite de cet apprentissage la présentation du stimulus sonore induit un ensemble de manifestations comportementales de peur telles qu’une immobilisation des animaux. Grâce à l’utilisation de techniques pharmacologiques et optogénétiques très novatrices, les chercheurs ont mis en évidence que les noyaux central et médian de l’amygdale centrale étaient différentiellement impliqués dans l’apprentissage et la manifestation comportementale des réponses de peur (cf. schéma). En effet, en inactivant la partie latérale du noyau central de l’amygdale les chercheurs ont pu montrer que les animaux n’apprenaient plus l’association entre le son et l’évènement désagréable. Au contraire, l’inactivation de la partie médiane de ce noyau ne perturbait pas l’apprentissage de la peur mais ne permettait plus aux animaux une manifestation comportementale de la peur, c’est à dire une immobilisation.
Dans une deuxième étape, l’enregistrement en temps réel de l’activité des neurones de l’amygdale centrale latérale et médiane grâce à des techniques électrophysiologiques uniques a permis au chercheurs d’identifier au sein de ces structures quels étaient les neurones spécifiquement impliqués dans l’apprentissage et la manifestation comportementale des réponses de peur.
Ces neurones sont des cellules inhibitrices qui font partie de circuits neuronaux très organisés et fortement interconnectés et dont les modifications d’activité permettent la sélection des réponses comportementales de peur pertinentes en fonction de la situation environnementale.