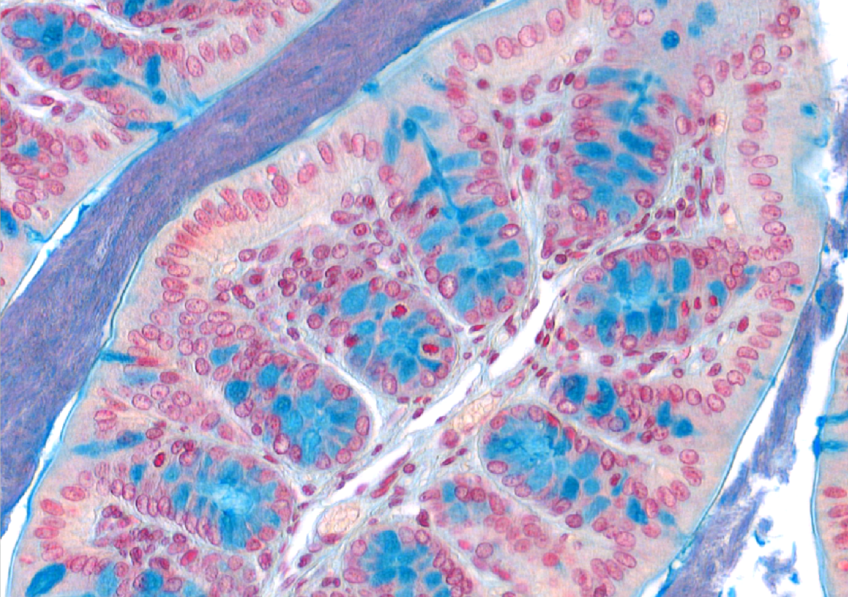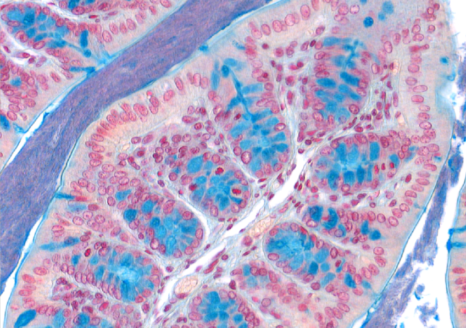
 Macrophages colonisés par Mycobacterium tuberculosis © Denis Fenistein-Priscille Brodin (Inserm)
Macrophages colonisés par Mycobacterium tuberculosis © Denis Fenistein-Priscille Brodin (Inserm)
L’équipe de recherche « Génétique Humaine des Maladies Infectieuses » de l’Université Paris Cité, de l’Inserm et de l’AP-HP, dirigée par le Pr Jean-Laurent Casanova et le Dr Laurent Abel, au sein de l’Institut Imagine, a identifié des variants rares du gène MCTS1 responsable d’une susceptibilité aux maladies mycobactériennes, comme la tuberculose. Ces travaux, publiés le 23 octobre dans la revue Cell, qui décrivent une nouvelle maladie génétique, confirment que l’interféron-gamma (IFN-g) – une protéine activant la réponse immunitaire – est le principal acteur de la réaction immunitaire dans ce type d’infection et que la protéine JAK2 est également nécessaire pour déclencher la réponse immunitaire en cas d’infection par une mycobactérie.
La tuberculose, causée par la bactérie Mycobacterium tuberculosis, est une maladie infectieuse mortelle qui a fait au moins un milliard de morts au cours des 2,000 dernières années et fait encore 1,5 millions de morts chaque année dans le monde. Pour 90% des personnes infectées, l’infection par M. tuberculosis est silencieuse ou bénigne. Dans de très rares cas (1 personne sur 50,000 environ), une défaillance de la réponse immunitaire peut être révélée par une infection sévère voire fatale, suite à l’injection du vaccin contre la tuberculose (dit vaccin BCG ou Bacille de Calmette et Guérin), qui contient pourtant une forme atténuée de la bactérie.
Jonathan Bohlen, post-doctorant, sous la direction du Pr Jean-Laurent Casanova, du Dr Qian Zhang et du Dr Jacinta Bustamante, a travaillé à l’identification des facteurs génétiques d’un groupe de maladies génétiques rares, prédisposant aux infections à des mycobactéries (comme celle de la tuberculose), et au bacille vaccinal de Calmette et Guérin (BCG). Ce groupe de maladies, dit MSMD (pour « Mendelian Susceptibility to mycobacterial diseases » en anglais) est présent à une fréquence plus élevée chez les hommes que chez les femmes.
Lors de ses travaux, J. Bohlen a mis en évidence des variants rares du gène MCTS1, situé sur le chromosome X, chez plusieurs garçons atteints de MSMD (notamment avec une atteinte osseuse) et provenant de plusieurs continents. Ces mutations du gène MCTS1 empêchent la production de la protéine du même nom, ce qui va altérer le fonctionnement des ribosomes, structures intracellulaires responsables de la production des protéines. En cas de mutation de MCTS1, comme observé chez les patients MSMD, le dysfonctionnement des ribosomes impacte la fabrication de certaines protéines spécifiques, comme la protéine JAK2 que l’équipe a identifiée et détectée en quantité plus faible chez ces mêmes patients. Le rôle de JAK2 étant d’activer la synthèse de l’interferon-g (IFN-g), une autre protéine impliquée dans l’activation de la réaction immunitaire face aux infections mycobactériennes, les patients présentant une mutation du gène MCTS1 sont donc plus susceptibles aux infections de ce type.
Ainsi, le déficit de MCST1 impacte négativement la production d’IFN-γ, ce qui entraîne une diminution de la réponse immunitaire face à une infection mycobactérienne, comme la tuberculose, chez ces patients. Ces travaux établissent donc une meilleure compréhension des mécanismes entre les mutations du gène MCTS1 et la réaction immunitaire anti-mycobactérienne.
De manière remarquable, une approche génétique à l’échelle du génome a permis d’éclaircir les mécanismes de réaction immunitaire face à une infection par des mycobactéries, dont le rôle de IFN-γ comme facteur immunitaire anti-mycobactérien. Dans l’ensemble, cette étude révèle un lien mécanistique surprenant entre la déficience d’un mécanisme biochimique général, et une prédisposition sélective aux infections à mycobactéries.
Ce travail marque une nouvelle avancée diagnostique et thérapeutique pour ce groupe de maladies infectieuses, avec l’identification d’un nouveau facteur génétique de prédisposition aux infections mycobactériennes.