crédit : ©Inserm
Une équipe de chercheurs de l’Inserm dirigée par Pierre Szepetowski (INMED, « Institut de Neurobiologie de la Méditerranée » Unité mixte Inserm/ Université d’Aix-Marseille) vient d’identifier un gène dont les mutations sont responsables d’un spectre large d’épilepsies et encéphalopathies épileptiques de l’enfant avec troubles du langage.
Ces travaux sont publiés dans la revue Nature Genetics.
Une crise d’épilepsie est liée à une activité excessive, soudaine et passagère d’un groupe de neurones. Elle se traduit par des manifestations cliniques paroxystiques (par exemple, les convulsions). Normalement, l’épilepsie n’altère pas les capacités cognitives. Toutefois, dans certaines formes appelées encéphalopathies épileptiques, la composante épileptique peut entraîner ou aggraver des troubles cognitifs et comportementaux sévères (déficience intellectuelle, trouble du langage, régression autistique, etc.). Elles se distinguent en cela des épilepsies « classiques ».
L’équipe et le réseau de chercheurs dirigés par Pierre Szepetowski tentent de mieux cerner ces relations entre les épilepsies et les nombreux autres troubles auxquels elles peuvent être liées : troubles autistiques, problèmes cognitifs, troubles du langage et de la parole, dyslexie, troubles du mouvement volontaire, migraines…
Jusqu’à présent, l’origine, débattue depuis plus de cinquante ans dans le monde médical et scientifique, de trois formes rares d’épilepsies et encéphalopathies épileptiques (épilepsie/aphasie « acquise », syndrome des pointes ondes continues du sommeil, et épilepsie Rolandique avec troubles de production du langage articulé), restait inconnue.
Grâce à une large analyse génétique, les chercheurs, dans le cadre d’un réseau étendu d’épileptologues et scientifiques associant différents centres hospitaliers et de recherche[1] viennent de montrer que 20 % de ces épilepsies souvent associées à des troubles du langage, ont une origine génétique commune.
Dans toutes ces formes, le gène GRIN2A codant pour un récepteur du glumatate, un neurotransmetteur crucial du cerveau, est muté.
Pour Pierre Szepetowski, ce nouvel éclairage montre que « ces trois syndromes peuvent être vus comme des expressions cliniques différentes d’une seule et même pathologie à la croisée des chemins entre l’épilepsie, les troubles du langage et les désordres cognitif et comportemental. »
L’identification de GRIN2A comme étant un gène majeur responsable de ces encéphalopathies épileptiques fournit des premières indications cruciales pour comprendre dans le futur les mécanismes sous-jacents.
« Ces encéphalopathies débutent généralement autour de 4-5 ans, après une période de développement normal. L’évolution en est très variable et imprévisible. L’identification d’une première cause majeure, va permettre de mieux expliquer la survenue de la maladie aux parents, notamment dans le cadre du conseil génétique ; on peut aussi espérer voir se mettre en place dans l’avenir, une fois les mécanismes mieux compris, des stratégies thérapeutiques précoces, qui seront cruciales pour améliorer un pronostic lié aux déficits neuropsychologiques associés. », conclut Pierre Szepetowski.
Prévenir in utero l’apparition de futures épilepsies ?
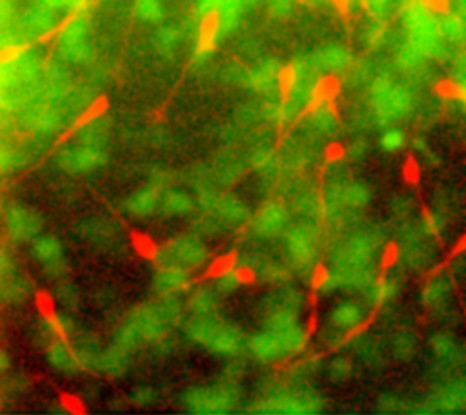
Dès la grossesse, des défauts dans le développement du cortex cérébral du futur bébé peuvent être à l’origine de l’apparition ultérieure de crises d’épilepsie. L’équipe de Pierre Szepetowski vient de montrer que l’absence d’une protéine, Srpx2, perturbe la migration neuronale dans le cerveau de rat en développement. Mieux, ils ont réussi à contrecarrer ces défauts et leurs conséquences épileptiques post-natales chez le rat grâce à l’administration maternelle de Tubacine, un produit capable de modifier le fonctionnement des tubulines – des molécules essentielles pour l’architecture des neurones et pour leur migration.
Ces travaux publiés début juillet dans Brain semblent constituer un premier pas montrant qu’il serait théoriquement envisageable d’empêcher in utero l’apparition future de certaines épilepsies.
[1] Lyon, Strasbourg, Reims et Marseille notamment