Les travaux réalisés par les équipes de Benoit Schneider et Odile Kellermann (Unité Inserm 747 « Cellules Souches, Signalisation et Prions », Université Paris Descartes) ainsi que de Jean-Marie Launay (Unité Inserm 942 Hôpital Lariboisière et fondation FondaMental), publiés cette semaine dans Nature Medicine, mettent au jour une enzyme, la kinase PDK1, impliquée dans l’accumulation, dans les neurones, des protéines pathologiques caractéristiques des maladies à prions et de la maladie d’Alzheimer. Les chercheurs démontrent que le blocage pharmacologique de cette enzyme exerce un effet bénéfique contre ces pathologies.
Le détail de ces travaux est publié dans la revue Nature Medicine
Les maladies à prions (maladie de Creutzfeld-Jakob chez l’homme) et la maladie d’Alzheimer sont associées à l’accumulation dans le cerveau de protéines anormales : la protéine prion scrapie (PrPSc) dans le cas des maladies à prions et les peptides amyloïdes Ab dans la maladie d’Alzheimer. La PrPSc et les peptides Ab exercent un effet toxique dans le cerveau en provoquant la mort des neurones, à l’origine des principaux signes cliniques caractéristiques de ces maladies.
Depuis quelques années, il est connu que la production des protéines pathologiques PrPSc et Ab40/42 a pour origine un défaut du clivage physiologique de la protéine prion entière non pathologique (PrPC) ou de la protéine précurseur des peptides amyloïdes (APP). Cependant, on ne savait pas, jusqu’alors, expliquer pourquoi ce clivage, qui normalement protège les neurones, est altéré dans les maladies à prions et la maladie d’Alzheimer.
Aujourd’hui, les travaux menés par Benoit Schneider, chercheur CNRS de l’Unité Inserm 747 « Cellules Souches, Signalisation et Prions », Université Paris Descartes et Jean-Marie Launay (Unité Inserm 942, Hôpital Lariboisière) en collaboration avec d’autres équipes françaises travaillant sur les prions viennent d’identifier une chaîne de réactions qui bloque le clivage bénéfique de la PrPC et APP par l’alpha-sécrétase TACE (acronyme pour TNFa Converting Enzyme). Les chercheurs dévoilent comment la dérégulation de TACE contribue à la neurodégénérescence en provoquant l’accumulation des protéines pathologiques PrPSc et Ab40/42 et en exacerbant la sensibilité des neurones à l’inflammation.
Dans des conditions physiologiques normales, TACE est présente à la surface des neurones, où elle exerce son activité de clivage à la fois vis-à-vis de la PrPC, de APP et des récepteurs au facteur inflammatoire TNFa (TNFR), ce qui limite considérablement la production des protéines pathologiques PrPSc et Ab et protège les neurones des effets délétères induits par TNFa.
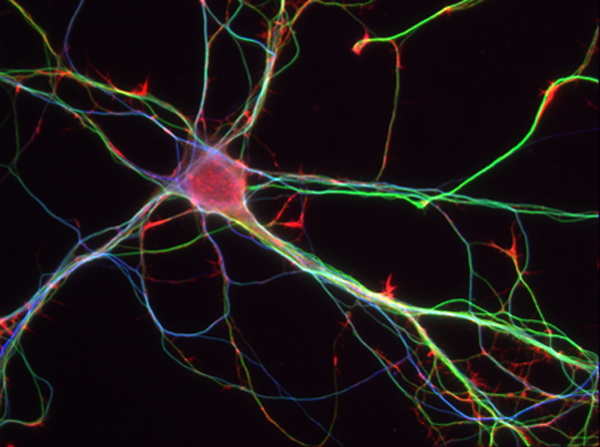
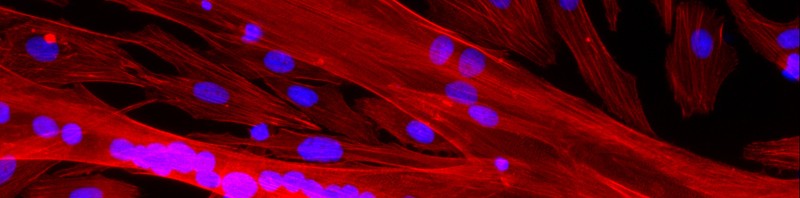
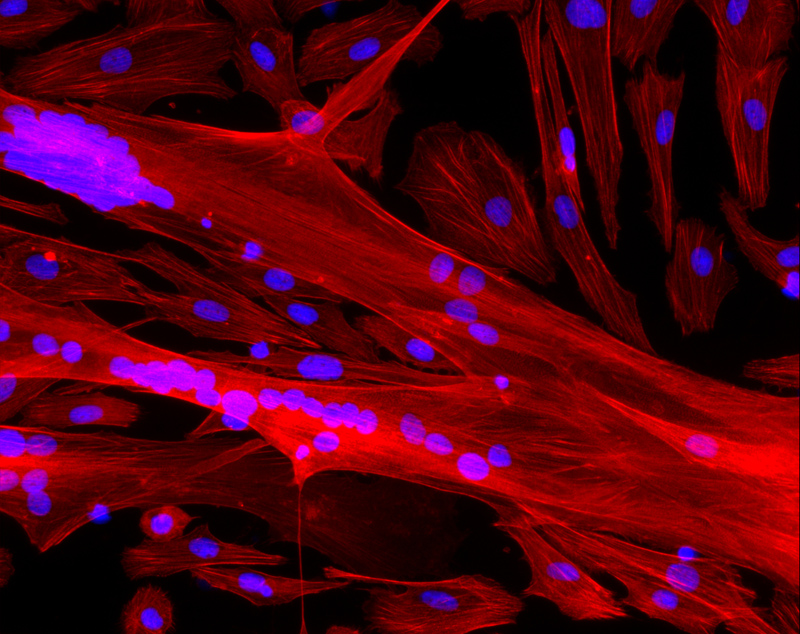
Neurones d’Hippocampes de souris à 7 jours – © Inserm/L.Peris
Dans les neurones infectés par les prions pathogènes comme dans les neurones « Alzheimer », la protéase TACE n’est plus localisée à la surface cellulaire, mais est séquestrée à l’intérieur des neurones. Cette internalisation empêche TACE de cliver ses substrats et d’exercer son activité neuroprotectrice. Les chercheurs dévoilent pour la première fois que la kinase PDK1 joue un rôle clé dans le contrôle de la localisation de TACE. La suractivation de PDK1 est responsable de la séquestration de TACE dans les neurones malades (infectés par les prions et Alzheimer) comme dans le cerveau des patients atteints de la maladie d’Alzheimer.
Le blocage pharmacologique de PDK1 renvoie TACE à la surface des neurones et restaure son rôle neuroprotecteur. L’inhibition de PDK1 parvient à protéger les neurones de la neurodégénérescence en restaurant les clivages physiologiques de la PrPC, APP et TNFR par TACE.

© Benoit Schneider & Mathéa Pietri, Août 2013.
« Grâce à nos travaux sur l’infection par les prions, nous avons pu identifier PDK1 comme une nouvelle cible thérapeutique non seulement pour la maladie de Creutzfeld-Jakob mais aussi pour la maladie d’Alzheimer »
expliquent les chercheurs.L’action de PDK1 sur TACE a été mise en évidence in vitro sur une lignée neuronale et des cultures de neurones provenant du cerveau de souris après infection par les prions, puis in vivo sur des modèles animaux. Un traitement par un inhibiteur pharmacologique de PDK1 permet d’atténuer les déficits moteurs et de prolonger la durée de vie des souris infectées par les prions pathogènes. Ensuite, en exploitant trois modèles de souris « Alzheimer », les chercheurs ont montré que ce traitement améliore aussi les troubles cognitifs des animaux et identifient PDK1 comme cible thérapeutique pour la maladie d’Alzheimer.
« Etant donné la rareté et l’efficacité réduite des traitements disponibles pour lutter contre les maladies à prions et la maladie d’Alzheimer, ces résultats offrent de nouvelles perspectives thérapeutiques pour le traitement de ces maladies neurodégénératives », concluent les chercheurs.
Ces travaux apportent un nouvel éclairage sur les mécanismes de neurodégénérescence induits par l’accumulation de protéines anormales. L’enjeu est maintenant de comprendre comment ces protéines toxiques conduisent à la dérégulation de PDK1, ce qui permettra d’identifier d’autres acteurs, c’est-à-dire de nouvelles cibles thérapeutiques potentielles, qui contribuent à la dégénérescence neuronale.

 © M Sieweke /Inserm
© M Sieweke /Inserm