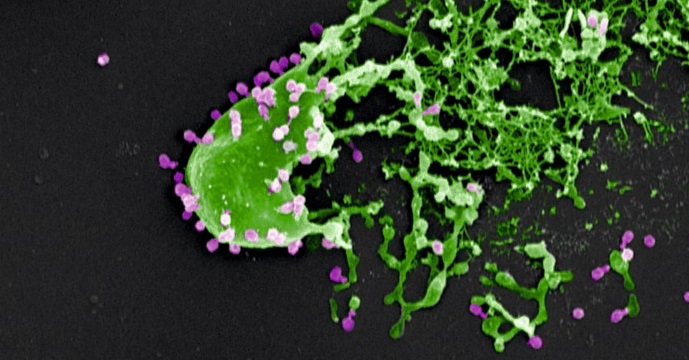
Bactéries (en vert) assaillies et détruites par des bactériophages (en violet). Cliché de microscopie électronique gracieusement fourni par M. Rohde and C. Rohde (Helmholtz Centre for Infection Research, Braunschweig/Leibniz Institute DSMZ, Braunschweig, Germany) et colorisé par Dwayne Roach (Institut Pasteur). © M. Rohde and C. Rohde.
Deux équipes de l’Institut Pasteur, en collaboration avec des chercheurs de l’Inserm et du Georgia Institute of Technology aux Etats-Unis, viennent de démontrer que, pour garantir la bonne efficacité d’une phagothérapie lors d’une infection bactérienne in vivo, l’action des bactériophages doit s’appuyer sur celle du système immunitaire de l’hôte. Cette synergie repose notamment sur l’action clé des cellules immunitaires neutrophiles. Cette découverte permet de mieux comprendre l’action thérapeutique des bactériophages dans le traitement de certaines infections bactériennes. Ces travaux sont publiés dans la revue Cell Host and Microbe le 12 Juillet 2017.
La phagothérapie repose sur l’utilisation de bactériophages (ou « phages ») pour traiter les infections bactériennes. Les phages sont des virus s’attaquant spécifiquement aux bactéries ; ils sont inoffensifs pour l’homme. Le recours à cette stratégie thérapeutique, conceptualisée il y a 100 ans, a connu un net recul dans le monde occidental, suite au développement des antibiotiques. Cependant, alors que le nombre d’infections causées par des bactéries résistantes aux antibiotiques augmente de façon alarmante, la phagothérapie connait actuellement un regain d’intérêt, notamment en Europe.
Jusqu’à présent, les données scientifiques n’étaient pas suffisantes pour comprendre le fonctionnement de la phagothérapie in vivo. En effet, la plupart des études menées in vitro avaient déjà prouvé que les phages tuent les bactéries qu’ils ciblent spécifiquement, mais aucune de ces études n’avait pu prendre en compte l’importance de la réaction de l’hôte face à cette activité.
Deux équipes de l’Institut Pasteur – le groupe Interactions bactériophages/bactéries chez l’animal de Laurent Debarbieux et l’unité Immunité innée dirigée par James Di Santo (Inserm U1223) -, en collaboration avec l’équipe de Joshua Weitz au Georgia Institute of Technology (Atlanta, Etats-Unis), viennent de démontrer l’importance du statut immunitaire du patient dans les chances de réussite d’une phagothérapie. Pour ce faire, ils ont mené une double approche originale en combinant un modèle animal et une modélisation mathématique.
Afin d’évaluer l’efficacité d’un traitement par une seule espèce de phages, les chercheurs se sont penchés sur la bactérie Pseudomonas aeruginosa qui est impliquée dans des infections respiratoires comme les pneumonies. Cette bactérie, résistante aux carbapénèmes, les « antibiotiques de la dernière chance », a été classée par l’OMS parmi les quatre plus menaçantes au niveau mondial pour des phénomènes de résistance aux antibiotiques.
Les chercheurs ont ainsi pu montrer que, chez les animaux avec un système immunitaire sain (dits « immunocompétents »), le traitement par phagothérapie est efficace. Le système immunitaire inné, rapidement mobilisable, et les phages agissent, dans un premier temps, en parallèle pour lutter contre l’infection. Puis, au bout de 24 à 48 heures, certaines bactéries deviennent naturellement résistantes aux phages qui ne peuvent plus assurer leur rôle. Le système immunitaire inné prend alors en charge la destruction de ces bactéries. Parmi les cellules immunitaires impliquées, les polynucléaires neutrophiles (des globules blancs provenant de la moelle osseuse) tiennent une place prépondérante.
Parallèlement, les simulations in silico ont permis de démontrer que la réponse innée doit assurer entre 20% et 50% de la destruction des bactéries afin que le traitement par phagothérapie soit efficace, et ce en l’absence ou bien en présence de phénomènes de résistance aux phages. Ainsi, sur le modèle étudié, les chercheurs ont prouvé qu’en aucun cas les phages seuls peuvent éradiquer une infection à P. aeruginosa.
Ces résultats sont d’autant plus importants qu’ils indiquent que les traitements par phagothérapie devraient prendre en compte le statut immunitaire des patients. Comme l’explique Laurent Debarbieux, « en termes de conséquences cliniques, il faudra probablement envisager la sélection des patients susceptibles de bénéficier d’un tel traitement. En effet, la phagothérapie pourrait ne pas être appropriée ou recommandée pour des personnes en situation d’immunodéficience sévère ».
Les chercheurs entendent maintenant décrypter précisément les voies immunitaires impliquées et les mécanismes sous-jacents. En parallèle, des essais cliniques sont en cours, notamment l’essai Phagoburn financé par le 7ème programme cadre de l’Union européenne, sur des infections cutanées chez de grands brûlés.