©2019 Flore Avram/Inserm
Aujourd’hui, environ un couple sur 8 consulte pour des difficultés à procréer. Les raisons sont probablement liées au fait que les projets parentaux ont lieu plus tardivement qu’autrefois, ou encore que les couples consultent plus facilement en mettant de côté les tabous liés à l’infertilité. Ainsi, l’infertilité est devenue un problème de santé publique et la communauté scientifique se mobilise.
Où en sont les recherches sur ces questions au cœur des problématiques sociétales actuelles ? Quel est leur transfert possible vers la clinique ? Les axes de recherche sur les troubles de la fertilité sont nombreux. L’objet de ce point presse n’est pas de les aborder de manière exhaustive mais d’évoquer les secteurs sur lesquels la recherche avance.
Quand la recherche avance, c’est la santé de tous qui progresse.
- Recherche contre l’infertilité
On parle d’infertilité au sein d’un couple lorsque celui-ci n’a pas réussi à avoir un enfant de manière naturelle après 12 mois de tentative. Cette définition englobe des situations de stérilité totale, sans espoir de conception naturelle, et une majorité de cas d’hypofertilité, c’est-à-dire de couples ayant des chances réduites – mais non nulles – d’obtenir une grossesse.
On classe ces infertilités en 4 catégories selon leur origine :
– 30 % sont d’origine féminine ;
– 30 % sont d’origine masculine. Chez l’homme, l’azoospermie, et l’oligospermie sont les deux principales causes d’infertilité identifiées à ce jour ;
– 30 % sont d’origine féminine et masculine, c’est-à-dire qu’elles sont liées à un déficit de fécondité qui touche les deux partenaires ;
– 10 % de cas sont inexpliqués.
Chez la femme, à l’exception des causes mécaniques tubaires – lorsque les trompes sont altérées ou bouchées (le plus souvent suite à une infection) – ou utérines, l’endométriose et les anomalies de l’ovulation sont les causes d’infertilité les plus fréquentes.
Parmi les anomalies de l’ovulation, on retrouve, entre autres, le syndrome des ovaires polykystiques (ce syndrome touche environ 10 % des femmes dans le monde), l’hyperprolactinémie et l’insuffisance ovarienne primaire (qui peut être aussi secondaire aux effets de la chimiothérapie).
La recherche actuelle vise, d’une part, à mieux comprendre les causes des infertilités et, d’autre part, à rechercher de nouveaux traitements ou prises en charge ayant pour objectif d’augmenter les chances de procréer.
1.1. Mieux comprendre les causes
Approche génétique
La recherche des causes génétiques de l’insuffisance ovarienne est un axe sur lequel de nombreux chercheurs travaillent. Certains gènes ne fonctionnent pas ou fonctionnent mal dans plusieurs troubles de la fertilité. L’un des domaines de recherche en pleine expansion, notamment du fait de l’amélioration des méthodes de criblage à haut débit, est la recherche de variants génétiques.
Le laboratoire Inserm de Nadine Binart travaille, par exemple, sur l’insuffisance ovarienne primaire (IOP) caractérisée par une incapacité de maturation des follicules ovariens ou une perte du pool des follicules de réserve. À partir de l’analyse de l’ADN de femmes atteintes d’IOP, les chercheurs travaillent à isoler dans leur patrimoine génétique les gènes impliqués/altérés. Cette approche aide à mieux comprendre les pathologies mais ne peut pas permettre de traiter spécifiquement ces femmes car, lorsque l’ovaire ne contient plus d’ovocytes, la stérilité est définitive. En revanche, une prise en charge préventive peut être déclenchée si l’anomalie génétique est retrouvée avant que le stock de follicules soit totalement épuisé – lors d’une enquête familiale par exemple. C’est le rôle de la recherche clinique qui permet de pallier ces maladies lorsque des mutations sont identifiées dans des familles atteintes, d’informer les jeunes patientes sur le risque de voir leur ovaire s’appauvrir en ovocytes au fil du temps et éventuellement de mettre en place des techniques de préservation de la fertilité.
Approche hormonale : exemple de la kisspeptine et de la prolactine
Il est bien établi que l’allaitement entraîne à la fois une augmentation de la sécrétion de prolactine (PRL) par l’hypophyse et inhibe les capacités d’une femme à ovuler. Ceci empêche la survenue d’une nouvelle grossesse. Il existe des situations pathologiques où la PRL augmente : c’est le cas des tumeurs situées sur l’hypophyse sécrétant cette hormone. Ces hyperprolactinémies, responsables de troubles des règles et d’infertilité, sont une cause majeure d’anovulation. L’équipe Inserm de Jacques Young et de Nadine Binart a permis, en 2011, de décortiquer le mécanisme sous-jacent du blocage du fonctionnement ovarien. Les chercheurs ont démontré, en utilisant un modèle de souris de la maladie, que la PRL inhibe la sécrétion d’une neuro-hormone appelée kisspeptine. Or, cette kisspeptine est le point de départ de toute la cascade hormonale responsable de la cyclicité ovarienne. Dans un modèle de souris, l’administration de kisspeptine a permis de rétablir le fonctionnement cyclique des ovaires malgré l’hyperprolactinémie.
Cette découverte physiopathologique explique pour la première fois le lien entre l’infertilité et l’hyperprolactinémie et permet une ouverture thérapeutique originale. La validation de ce concept chez la femme vient d’être réalisée[1] ce qui permettra de proposer une alternative thérapeutique en cas de résistance aux médicaments actuels.
1.2. Préserver la fertilité : axes de recherche et derniers résultats
Depuis plusieurs années, des consultations spécialisées dites d’oncofertilité se sont largement développées et doivent maintenant faire partie intégrante du parcours de soin des patientes jeunes atteintes de cancer. Plusieurs techniques dites de préservation de la fertilité visant à cryoconserver des gamètes ou à préserver les capacités reproductives sont aujourd’hui disponibles et d’autres sont en cours de développement. En France, ces démarches s’inscrivent, depuis 1994, dans les différentes lois de bioéthique. L’article L.2141 11, modifié par la loi 2011-814 du 7 juillet 2011 prévoit que « Toute personne dont la prise en charge médicale est susceptible d’altérer la fertilité, ou dont la fertilité risque d’être prématurément altérée, peut bénéficier du recueil et de la conservation de ses gamètes ou de ses tissus germinaux, en vue de la réalisation ultérieure, à son bénéfice, d’une assistance médicale à la procréation, ou en vue de la préservation et de la restauration de sa fertilité ». Par ailleurs, le plan Cancer 2014-2019 a intégré des mesures de préservation de la fertilité stipulant que « l’accès aux traitements du cancer et en particulier aux traitements innovants, doit être garanti à tous les malades ».
Améliorer la conservation des gamètes
Plusieurs techniques permettant de cryoconserver des gamètes des femmes sont aujourd’hui disponibles. La congélation d’ovocytes matures ou d’embryons obtenus à partir de ces ovocytes représente la méthode de référence. Cependant, elle ne peut être réalisée chez les jeunes filles prépubères, lorsque le traitement doit être débuté en urgence. Elle peut également être problématique dans le cadre de pathologies hormono-sensibles. Aussi, d’autres techniques, bien qu’encore considérées comme expérimentales, peuvent être proposées dans ces situations.
Actuellement l’amélioration des méthodes disponibles et le développement de nouvelles stratégies sont un enjeu majeur en oncofertilité. C’est l’objet d’un des axes de recherche de l’équipe Inserm de Nadine Binard et Charlotte Sonigo en collaboration avec le Pr Michael Grynberg.
Utiliser l’hormone anti-müllérienne
La chimiothérapie fait baisser la fertilité en exerçant une toxicité directe sur les ovaires. Couramment utilisé dans le traitement du cancer, le cyclophosphamide provoque une destruction massive des cellules germinales contenues dans les follicules ovariens. Les chercheurs viennent de montrer, dans un modèle de souris, qu’un traitement par l’hormone anti-müllérienne, normalement secrétée par les ovaires, permettait de limiter la réduction du stock de follicules lors d’une chimiothérapie. L’hormone anti-müllérienne constitue ainsi une nouvelle promesse pour préserver la fertilité.
1.3. L’apport des nouvelles technologies : L’intelligence artificielle au service de la recherche en reproduction
Le stock des cellules germinales, contenues dans les follicules, constitue la réserve ovarienne. L’évaluation de la quantité de ces cellules germinales est couramment utilisée pour comprendre la physiologie ovarienne ou mesurer l’impact de l’environnement sur les ovaires. La méthode de référence utilisée chez la souris est longue et fastidieuse. Les chercheurs de l’Inserm viennent de développer, avec une entreprise spécialisée dans l’intelligence artificielle, une méthode d’intelligence artificielle automatisée de comptage folliculaire par deep learning[2]. Ce nouvel outil sera disponible pour la communauté scientifique s’intéressant à la fertilité permettant un grand gain de temps et une meilleure reproductibilité des données.
- Recherche contre l’endométriose
L’endométriose est un syndrome complexe caractérisé par un processus inflammatoire chronique dû à la présence de tissu semblable à la muqueuse utérine en dehors de l’utérus. Cet « utérus ectopique » continue à fonctionner sous l’influence des hormones ovariennes provoquant chez certaines femmes de fortes douleurs et parfois une infertilité. En parallèle d’une médiatisation importante, notamment sous l’impulsion des associations de malades, la ministre de la Santé a annoncé un plan d’action pour renforcer la prise en charge de l’endométriose. Au niveau de la recherche, il existe une explosion des études sur le sujet depuis les 5 dernières années. Environ 1200 articles par an sont produits par les chercheurs du monde entier pour faire avancer les connaissances sur cette pathologie.
Qu’est ce que l’endométriose ?
©2019 Flore Avram/Inserm
- 1 femme sur 10 serait concernée par une forme d’endométriose
- les localisations des lésions d’endométriose sont hétérogènes
- le reflux de cellules de l’endomètre au moment des règles existe chez 90 % des femmes mais seules 10 % développent une pathologie
- 4 stades sont classiquement décrits pour la maladie en fonction de l’étendue des lésions et de leur profondeur ; cependant, il n’y a pas de corrélation entre les symptômes et la sévérité de la maladie
- on distingue 3 formes d’endométriose : l’endométriose superficielle ou péritonéale, l’endométriose ovarienne (ou kyste endométriosique, ou endométriome) et l’endométriose profonde.
2.1. Mieux comprendre les causes
L’approche épidémiologique
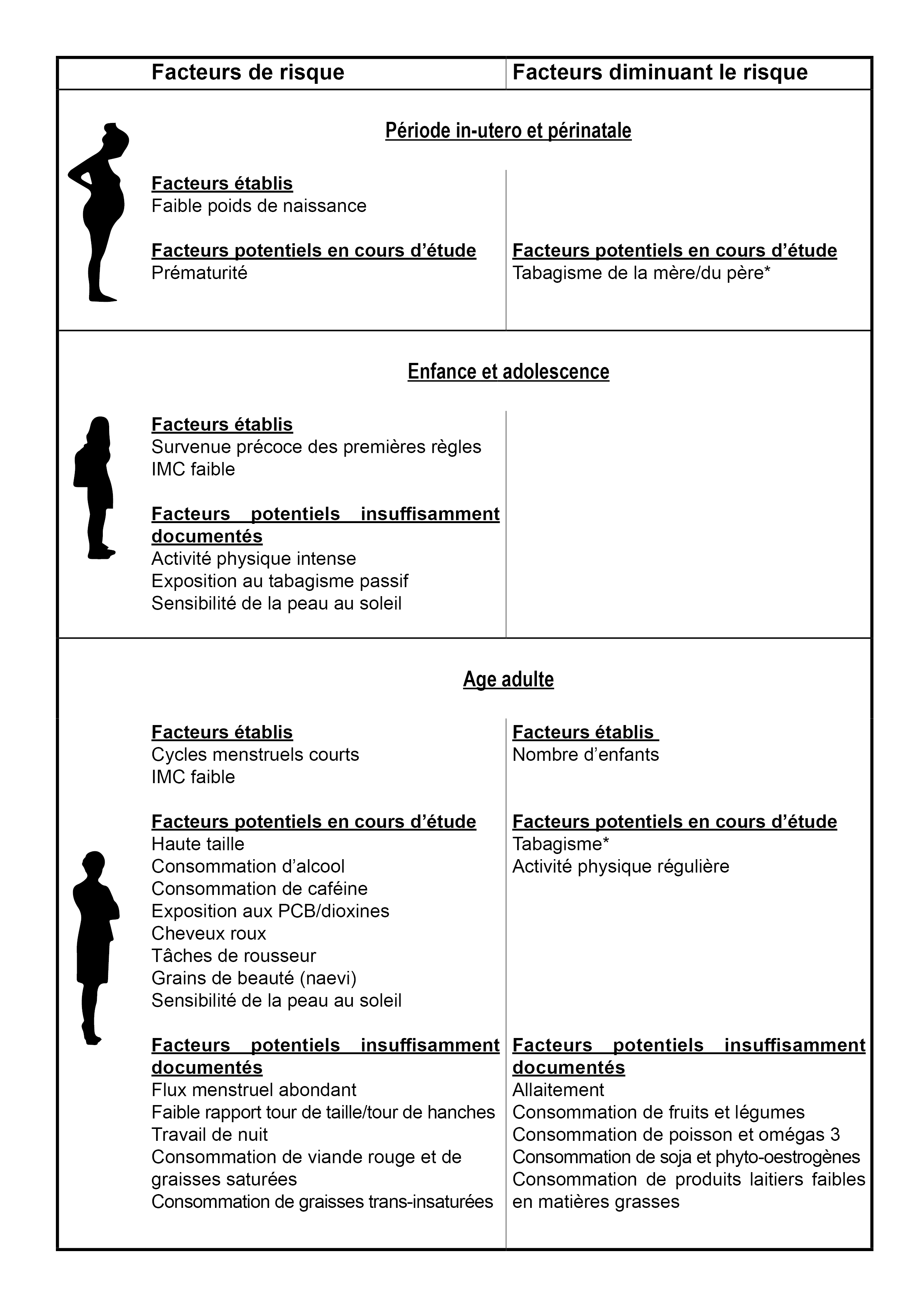
Aujourd’hui peu de choses sont connues sur les causes de l’endométriose, son évolution naturelle et les facteurs qui influencent sa progression. L’apport de la recherche en épidémiologie est primordial pour faire avancer ces connaissances. Seules quelques grandes cohortes épidémiologiques permettant d’explorer ces aspects existent à travers le monde. En termes d’exploration des facteurs de risque d’endométriose, la plus grande à ce jour est une cohorte de 116 430 infirmières américaines âgées de 25 à 42 ans en 1989. Parmi les facteurs de risques identifiés dans la littérature et confirmés dans cette cohorte : un faible poids de naissance, des menstruations précoces, un faible indice de masse corporel et des cycles menstruels courts (moins de 24 jours)[3]. Néanmoins, au-delà de ces facteurs, peu de connaissances sont disponibles sur les causes de la maladie, et son histoire naturelle est largement inconnue. Une revue de la littérature parue en août 2018 a permis de dresser le tableau suivant :

* L’association positive entre le tabagisme et la diminution du risque d’endométriose pourrait trouver une explication dans l’effet anti-oestrogénique du tabac. Cette observation viendrait confirmer l’intérêt thérapeutique des antiestrogènes pour lesquels il existe des médicaments beaucoup plus recommandables que la cigarette dont les effets nocifs sont largement documentés.
Afin de mieux comprendre cette maladie, plusieurs projets épidémiologiques voient le jour en France sous l’impulsion de l’équipe de Marina Kvaskoff, épidémiologiste et chercheuse à l’Inserm. Parmi ceux-ci, une cohorte de patientes dédiée à l’étude de l’endométriose vient d’être mise en place : la cohorte ComPaRe-Endométriose. L’objectif de l’équipe scientifique est d’atteindre un nombre suffisamment important de femmes incluses dans cette cohorte pour obtenir des résultats robustes aux nombreuses questions encore en suspens sur cette pathologie. En moins de 6 mois, déjà plus de 8000 femmes participent à l’étude. L’équipe vise à recruter 15 à 20 000 participantes ; un large appel à participation est lancé aux femmes atteintes d’endométriose ou d’adénomyose (endométriose restreinte au muscle de l’utérus) pour accélérer les recherches sur ces pathologies, en répondant simplement à des questionnaires en ligne sur leur vécu de la maladie (https://compare.aphp.fr/). Les premiers axes de recherche sont d’explorer l’histoire naturelle de la maladie (évolution des symptômes et des caractéristiques de la maladie au cours du temps), et d’identifier les facteurs déterminant sa progression et ceux menant à une meilleure réponse au traitement. L’étude permettra également de décrire les circonstances du diagnostic et les parcours de soin des patientes, et d’examiner l’impact de la maladie sur leur quotidien.
L’étude de l’endométriose fait également l’objet de projets au sein d’autres grandes cohortes françaises, comme la cohorte CONSTANCES, une étude prospective de 200 000 hommes et femmes (105 000 femmes) représentative de la population française. L’équipe de Marina Kvaskoff a développé un projet de recherche épidémiologique qui permettra de déterminer la prévalence et l’incidence de la maladie en France ainsi que d’explorer ses facteurs de risque dans cette cohorte. D’autres travaux sont en développement et seront menés à terme dans d’autres cohortes.
La piste de l’environnement
Plusieurs études épidémiologiques ont exploré les associations entre les produits chimiques organochlorés (solvants, pesticides, insecticides, fongicides…) et l’endométriose, mais les résultats ne sont pas uniformes. Une méta analyse française de 17 études[4] a été publiée en février 2019 pour essayer de tirer des résultats plus robustes. Le risque de développer une endométriose était de 1,65 plus élevé chez les femmes exposées aux dioxines ; 1,70 pour celles exposées aux polychlorobiphényles (PCB) et 1,23 pour les pesticides organochlorés. Bien qu’elles soient statistiquement significatives, ces estimations doivent être considérées avec prudence en raison de leur hétérogénéité notable entre les études et de la faible ampleur de l’effet estimé. Le niveau de preuve a été jugé » modéré » avec un risque sérieux de biais ce qui justifie la nécessité de mener d’autres recherches épidémiologiques bien conçues pour combler les lacunes persistantes des données.
L’approche génétique et épigénétique pour un dépistage précoce
Le dépistage de l’endométriose à des stades précoces et en l’absence de symptômes permettrait une meilleure prise en charge des patientes. Si l’héritabilité de l’endométriose a été évaluée à 50 %, elle est très complexe et manifestement très polygénique. De nombreux gènes candidats ont été étudiés de ce point de vue dans des analyses de prédisposition à la maladie. Les premiers résultats ont montré qu’il n’existe pas un gène de l’endométriose mais que l’existence de variants génétiques caractéristiques de la pathologie pourraient permettre de diagnostiquer celle-ci et d’améliorer la prise en charge des patientes En 2017, les efforts de la communauté internationale ont permis d’identifier au total 14 variants (situés dans les gènes WNT4, GREB1, ETAA1, IL1A, KDR, ID4, CDKN2B-AS1, VEZT, FN1, CCDC170, SYNE1, FSHB et dans les régions chromosomiques 7p15.2 et 7p12.3). Ces 14 gènes sont impliqués dans la prolifération et le cycle cellulaire, l’adhésion et la matrice extracellulaire et l’inflammation, ce qui fait sens en matière d’endométriose. Néanmoins, chacun des variants identifiés n’explique qu’une part limitée de la variation génétique dans l’endométriose. La combinaison d’allèles à risque chez une patiente pourrait dans l’avenir donner une probabilité d’être atteinte utilisable pour diagnostiquer les patientes et les classer en fonction du type d’endométriose et de sa gravité.
Par ailleurs, l’existence de marques épigénétiques spécifiques de l’endométriose pourrait aussi permettre en théorie le dépistage précoce. Les cellules endométriosiques présentent effectivement des anomalies épigénétiques spécifiques qui modifient l’expression des principaux facteurs de transcription. Toutefois, on ne sait pas comment les interactions entre les cellules épigénomiques défectueuses et les gènes mutés des cellules épithéliales contribuent à la pathogenèse de l’endométriose.
La piste des microARN
La génétique est cependant insuffisante pour rendre compte de l’endométriose dans sa complexité. Les gènes n’interviennent dans le phénotype qu’en tant qu’ils sont exprimés. La régulation de cette expression passe par des mécanismes moléculaires épigénétiques. De ce point de vue, la plupart des études porte sur la recherche de microARN qui « marqueraient » la maladie. Pour l’instant, plusieurs ont été identifiés dans le plasma des patientes, avec cependant une reproductibilité très mauvaise d’une équipe à l’autre. Par exemple, une étude publiée en 2013[5] identifie quatre miARN seulement (miR-199a, miR-122, miR145* et miR-542-3p) comme suffisant pour classer, avec très peu d’erreurs, les patientes. Néanmoins, la confirmation sur des cohortes indépendantes des résultats de cet article tarde à venir. Une explication possible est le fait que l’extraction des ARN circulants demeure très hétérogène d’une étude à l’autre, peut-être en relation avec les outils techniques utilisés lors de l’extraction. Dans l’avenir, de nouvelles approches plus exhaustives pourraient apporter des résultats plus homogènes.
L’approche cellulaire : le stress oxydatif
Plusieurs études ont montré une augmentation du stress oxydatif dans le sérum des femmes atteintes d’endométriose. Le stress oxydatif est un mécanisme très général induisant et causé par l’inflammation. Face une maladie douloureuse, comme l’endométriose, trouver des altérations liées au stress oxydatif n’est pas surprenant. Dans des modèles murins, un traitement par des antioxydants ( N acetyl cystéine) a permis la réduction des lésions endométriosiques.
Par ailleurs, par une approche menée par les chercheurs de l’Institut Cochin, parmi les cascades de gènes dérégulées dans la lésion d’endométriose, ils ont trouvé de nombreux gènes liés au métabolisme du glutathion. Ce tripeptide joue un rôle clef dans la détoxification du peroxyde d’hydrogène, molécule majeure du stress oxydatif. La dérégulation négative en particulier des gènes GCLM et GCLC cruciaux pour la synthèse du glutathion, pourrait expliquer un accroissement du stress oxydatif dans les lésions d’endométriose.
La piste de la défaillance immunitaire ?
La survie des cellules endométriosiques à l’extérieur de l’utérus pourrait être liée à un mauvais fonctionnement du système immunitaire duquel résulterait une inflammation chronique locale, et un échec de ll’élimination de ces cellules ectopiques. Les mécanismes immunitaires en jeu restent mal compris, mais plusieurs éléments pointent à une dérégulation des cellules du système immunitaire, tels que les macrophages et les lymphocytes B. Les macrophages sont des cellules de l’immunité innée participant à l’inflammation et à l’élimination des débris cellulaires. Il a été montré que les macrophages de patientes souffrant d’endométriose favorisaient la croissance des cellules endométriosiques in vitro. Les lymphocytes B sont des cellules de l’immunité adaptative, et sont responsables de la fabrication d’anticorps. Leur dérèglement dans l’endométriose est illustré par une activation importante et la présence d’auto-anticorps contre des antigènes de l’endomètre. D’autres cellules immunitaires sont également impliquées et une recherche active pour mieux comprendre leur implication est en cours.
2.2. Les traitements : axes de recherche et derniers résultats
Changer les modalités du diagnostic : vers la fin de la chirurgie
Avant d’envisager un traitement, la première étape est de réduire le temps de diagnostic de l’endométriose aujourd’hui estimé entre 7 et 10 ans après l’apparition des premiers symptômes. Pour cela, médecins et chercheurs travaillent à l’élaboration d’un score diagnostique, basé sur une dizaine de questions à partir desquelles le médecin pourra poser un diagnostic fiable à 85-90 %. Ce score pourra être complété si nécessaire par des examens d’imagerie qui, si ils sont réalisés et interprétés par des personnels médicaux formés, peuvent tout à fait renseigner le diagnostic d’endométriose.
Médecins et chercheurs s’accordent à dire qu’il contre-indiqué de pratiquer des chirurgies à visée diagnostiques pour l’endométriose.
Les 3 piliers du traitement
Le traitement médicamenteux, la chirurgie et l’assistance médicale à procréation (AMP) sont les 3 seules approches existantes pour traiter les symptômes de l’endométriose et ses éventuelles conséquences sur la fertilité. L’enjeu majeur – en l’absence de nouveaux traitements – est de bien comprendre quel rôle joue chacune des composantes de cet arsenal thérapeutique afin de l’utiliser à bon escient.
Le traitement médicamenteux repose sur le blocage des fonctions ovariennes pour créer une ménopause artificielle via des contraceptifs pris en continu. Ces traitements doivent être personnalisés et adaptés à chaque patiente (estroprogestatifs, progestatifs, analogues de la GnRH. Ils doivent être prescrits en première intention chez la femme sans désir de grossesse et ce afin de réduire les douleurs liées à cette pathologie.
Lors d’un projet de grossesse, l’AMP et la chirurgie peuvent être envisagées. Avant tout geste chirurgical, le recours à l’AMP doit être systématique afin de maximiser les chances de concevoir un enfant pour les couples qui le souhaitent. La chirurgie ne doit pas être utilisée chez des femmes sans projet d’enfant pour lesquelles le traitement médicamenteux fonctionne. La chirurgie de l’endométriose peut être très invasive et invalidante (résection de certaines parties du côlon, risque élevé d’abîmer la réserve ovarienne en cas de retrait de kyste ovarien etc.,) et, en ne traitant pas la cause, n’empêche pas la maladie de revenir. Là encore, médecins et chercheurs s’accordent sur le fait qu’une femme opérée jeune présente un risque important que les lésions endométriosiques reviennent et soient à nouveau problématiques au moment d’un éventuel souhait de grossesse.
Tout doit donc être fait pour que la chirurgie ne soit plus le traitement de référence de l’endométriose comme cela a été trop le cas dans le passé.
Aujourd’hui, certaines formes d’endométriose – notamment celles qui touchent les ovaires – sont une indication permettant aux femmes d’avoir accès aux différentes techniques de préservation de la fertilité.
L’apport des nouvelles technologies : l’exemple des ultrasons haute-fréquence
A Lyon, des équipes de cliniciens chercheurs sous l’impulsion du Pr Gil Dubernard (Hospices Civils de Lyon et unité Inserm 1032 LabTAU) ont mis au point un traitement de l’endométriose rectale à base d’ultrasons. Lorsque l’endométriose infiltre la paroi rectale, elle est responsable de douleurs rectales invalidantes qui peuvent altérer la qualité de vie. Après échec du traitement médical, une chirurgie est souvent proposée qui consiste à retirer une partie ou la totalité du rectum et nécessite parfois une dérivation digestive (anus artificiel) transitoire.
Une étude clinique de phase I menée sur 11 patientes a démontré en 2017 que les ultrasons de haute intensité peuvent être une alternative intéressante à la chirurgie. En effet, en quelques minutes, grâce à une sonde à ultrasons introduite par voie rectale, la lésion est « désensibilisée ». Afin de confirmer ces premiers résultats, un nouvel essai incluant 12 patientes s’est terminé le 1er avril 2019. L’analyse des données est en cours et sera bientôt disponible.
En parallèle et en collaboration avec la société EDAP-TMS (promotrice des essais cliniques), le laboratoire Inserm dédié aux ultrasons thérapeutiques dirigé par Cyril Lafon, le LabTAU (Université Claude Bernard Lyon 1/Inserm), travaille sur l’optimisation des conditions de délivrance des ultrasons (insonification) et l’amélioration de l’ergonomie de la sonde pour augmenter le nombre de patientes éligibles à ce nouveau traitement.
Cette thérapeutique innovante permettra très probablement de remplacer une bonne partie des chirurgies rectales réalisées pour cette maladie fonctionnelle qui guérit à la ménopause.
[1] Hypothalamic-Pituitary-Ovarian Axis Reactivation by Kisspeptin-10 in Hyperprolactinemic Women With Chronic Amenorrhea.
Millar RP, Sonigo C, Anderson RA, George J, Maione L, Brailly-Tabard S, Chanson P, Binart N, Young J.
[2] Sonigo C, Jankowski S, Yoo O, Trassard O, Bousquet N, Grynberg M, Beau I, Binart N. High-throughput ovarian follicle counting by an innovative deep learning approach. Sci Rep. 2018 Sep 10;8(1):13499. doi: 10.1038/s41598-018-31883-8.
[3] https://www.ncbi.nlm.nih.gov/pubmed/30017581
[4] https://www.ncbi.nlm.nih.gov/pubmed/30530163
[5] (Wang et al, JCEM , 2013)