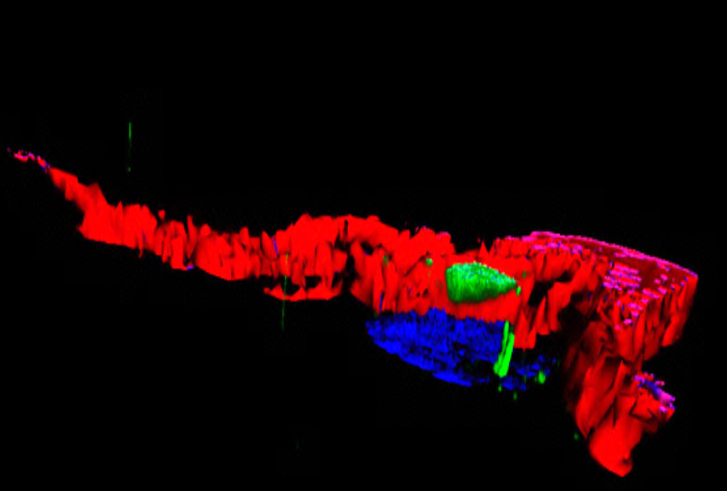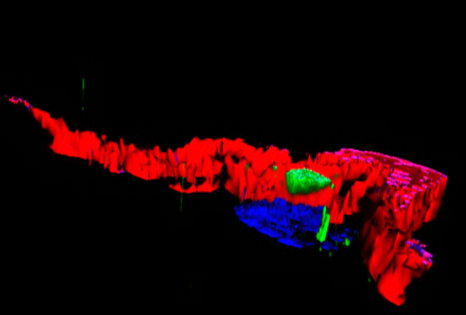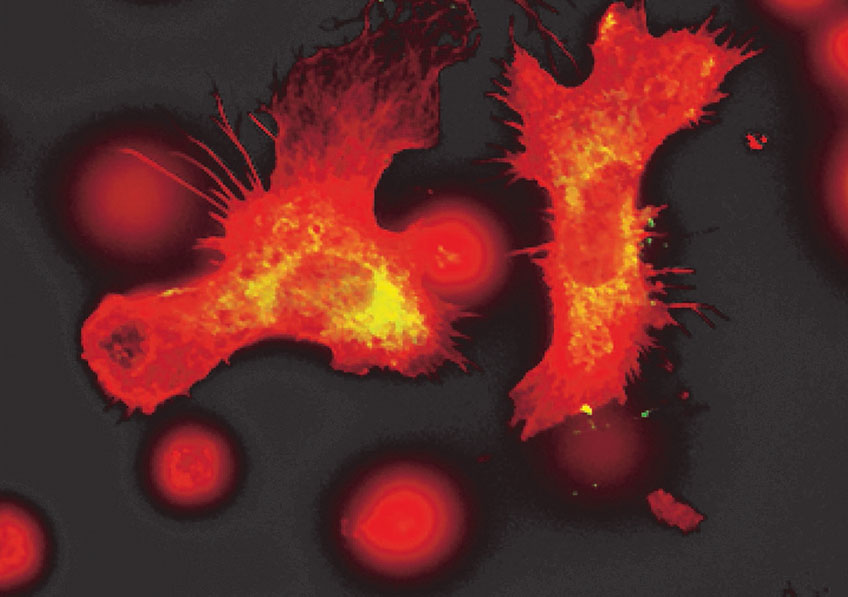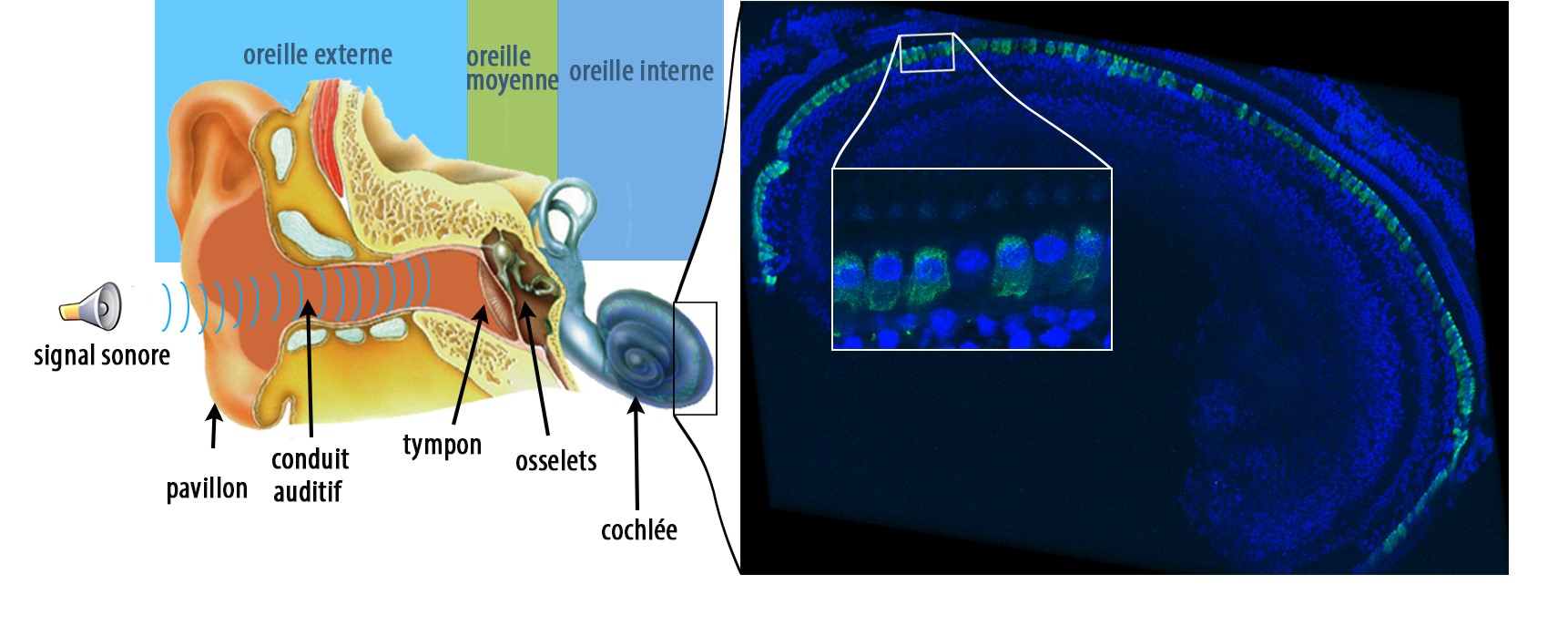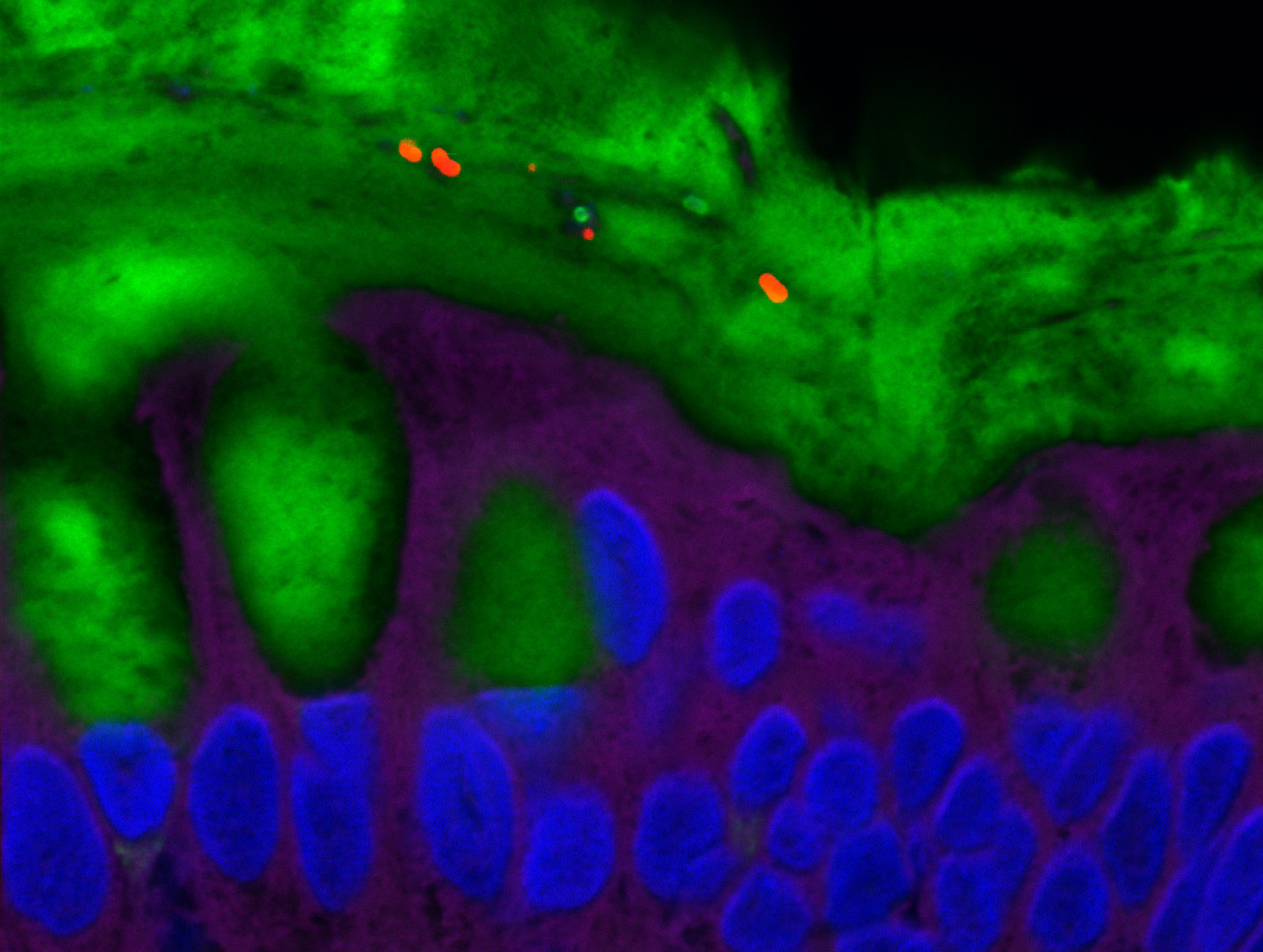
Certaines bactéries du microbiote intestinal, marquées en rouge, sont capables de pénétrer la couche de mucus normalement stérile et marquée en vert. © Benoit Chassaing
L’alimentation jouerait un rôle dans le déclenchement d’inflammations intestinales pouvant aboutir au développement de certaines pathologies, comme la maladie de Crohn. Des chercheurs de l’Inserm, du CNRS et de Université de Paris ont montré que les émulsifiants alimentaires présents dans de nombreux plats transformés pouvaient avoir un impact délétère sur certaines bactéries spécifiques du microbiote intestinal, conduisant à une inflammation chronique. Leurs résultats sont publiés dans Cell Reports.
La prévalence des maladies inflammatoires chroniques de l’intestin ne cesse d’augmenter dans tous les pays du monde. Près de 20 millions de personnes seraient concernées. Caractérisées par l’inflammation de la paroi d’une partie du tube digestif, ces pathologies regroupent notamment la maladie de Crohn et les rectocolites hémorragiques.
Plusieurs facteurs, à la fois génétiques et environnementaux, ont été mis en cause pour expliquer l’inflammation de l’intestin associée à ces maladies. Depuis plusieurs années, le chercheur Inserm Benoît Chassaing et son équipe à l’Institut Cochin (Inserm/CNRS/Université de Paris) s’intéressent au rôle de l’alimentation et notamment à l’impact de certains additifs alimentaires comme les émulsifiants.
Largement utilisés par l’industrie agroalimentaire dans de nombreux produits transformés, les émulsifiants[1] ont pour fonction d’en améliorer la texture et d’en prolonger la durée de conservation. Par exemple, des émulsifiants comme la lécithine et les polysorbates permettent de garantir la texture onctueuse des crèmes glacées industrielles et d’éviter qu’elles ne fondent trop rapidement une fois servies.
Dans différents travaux s’appuyant sur des modèles animaux, les scientifiques ont déjà montré que la consommation d’émulsifiants alimentaires altérait négativement le microbiote de manière à favoriser l’inflammation.
Par ailleurs, dans des modèles de souris dont le microbiote était composé d’une faible diversité de bactéries, les chercheurs ont observé que les animaux étaient protégés contre les effets négatifs de certains émulsifiants.Ils ont donc émis l’hypothèse que les émulsifiants impacteraient seulement certaines bactéries spécifiques, inoffensives dans des conditions « normales », mais ayant un potentiel pathogène. C’est seulement en présence d’agents émulsifiants que ces dernières seraient capables de favoriser le développement d’une inflammation intestinale chronique et de maladies associées.
E. coli comme un modèle
Dans le cadre de leur étude publiée dans Cell Reports, les chercheurs ont cette fois ci travaillé à partir de deux modèles de souris : l’un sans microbiote et l’autre avec un microbiote simple comportant seulement 8 espèces de bactérie. Ils les ont colonisés avec une souche de la bactérie Escherichia coli (les « bactéries AIEC ») associée à la maladie de Crohn.
Les chercheurs se sont intéressés aux effets de deux émulsifiants administrés suite à la colonisation des souris par les bactéries AIEC. Alors que la seule consommation d’agents émulsifiants était inoffensive chez ces animaux en l’absence de ces bactéries, ils ont constaté le développement d’une inflammation intestinale chronique et de dérégulations métaboliques lorsque ces dernières étaient présentes. Ainsi, le « couple » bactéries AIEC / agent émulsifiant était nécessaire et suffisant pour induire une inflammation intestinale chronique.
Des analyses supplémentaires ont révélé que lorsque ces bactéries étaient en contact avec les émulsifiants, elles sur-exprimaient des groupes de gènes qui augmentaient leur virulence et leur propension à induire l’inflammation. « Nous avons ainsi pu identifier un mécanisme par lequel les émulsifiants alimentaires peuvent favoriser l’inflammation intestinale chronique chez les personnes abritant certaines bactéries, telles que les bactéries AIEC, dans leur tractus digestif », souligne Benoît Chassaing qui a coordonné l’étude.
La prochaine étape consiste à lister l’ensemble des bactéries ayant les mêmes effets au contact de ces additifs alimentaires.
A plus long terme, des études pour identifier et stratifier les patients en fonction de la composition de leur microbiote et de leur risque d’inflammation pourraient être mises en place dans le but de faire de la prévention et de mettre en place des recommandations nutritionnelles personnalisées. Les personnes porteuses de microbiotes spécifiques, sensibles aux émulsifiants, pourraient en effet bénéficier de recommandations alimentaires ciblées.
« Et s’il est illusoire de penser que l’on pourra bannir les émulsifiants de notre alimentation, les modèles et les méthodologies que nous avons développés ici vont aussi nous permettre de tester l’action de plusieurs types d’agents émulsifiants sur le microbiote afin identifier ceux qui n’auraient pas d’effets délétères, et ainsi encourager leur usage », conclut Benoît Chassaing.
[1] Un émulsifiant est un composé qui a une affinité à la fois avec l’eau et avec l’huile et qui permet aux différentes phases d’un composé de rester mélangées