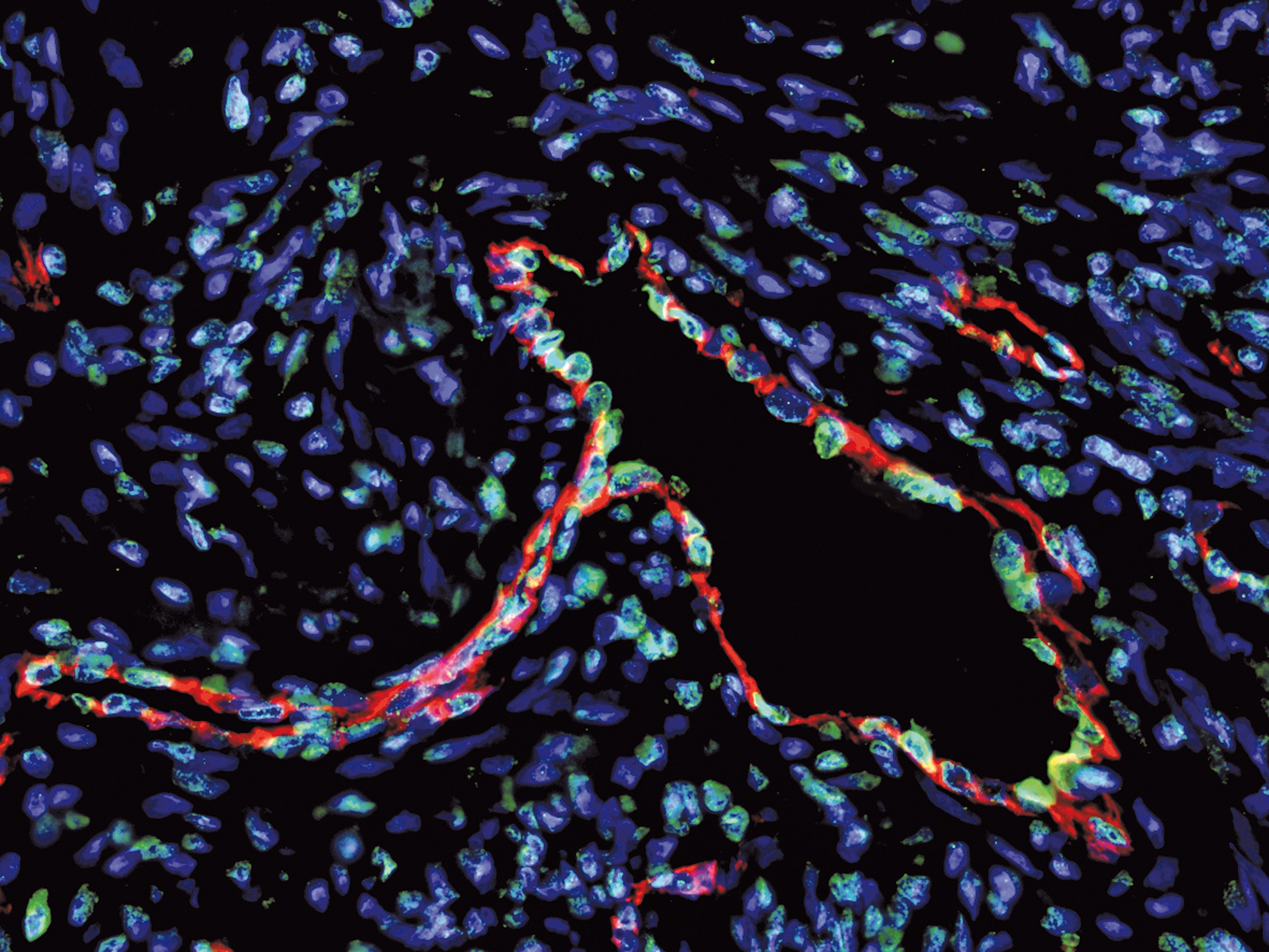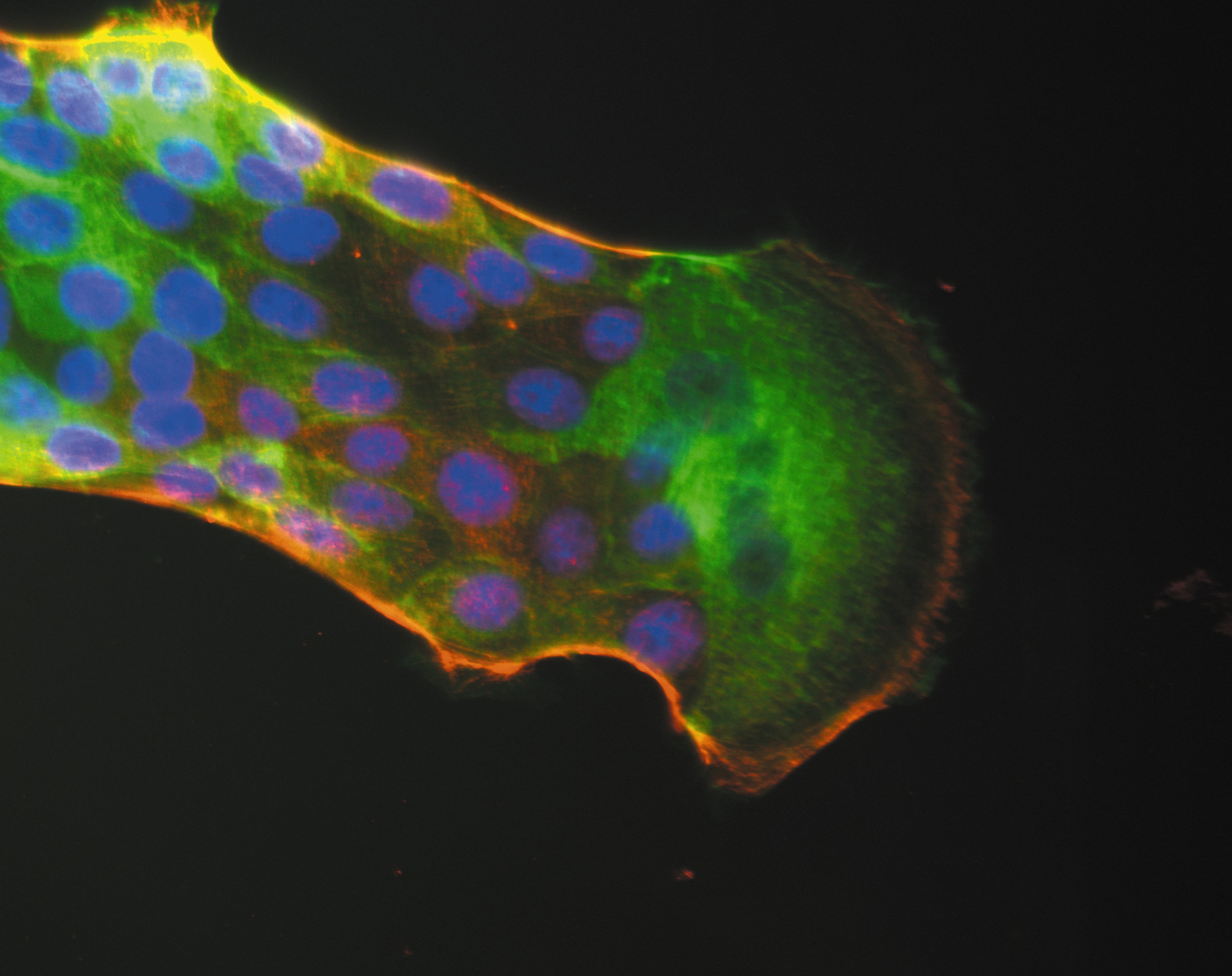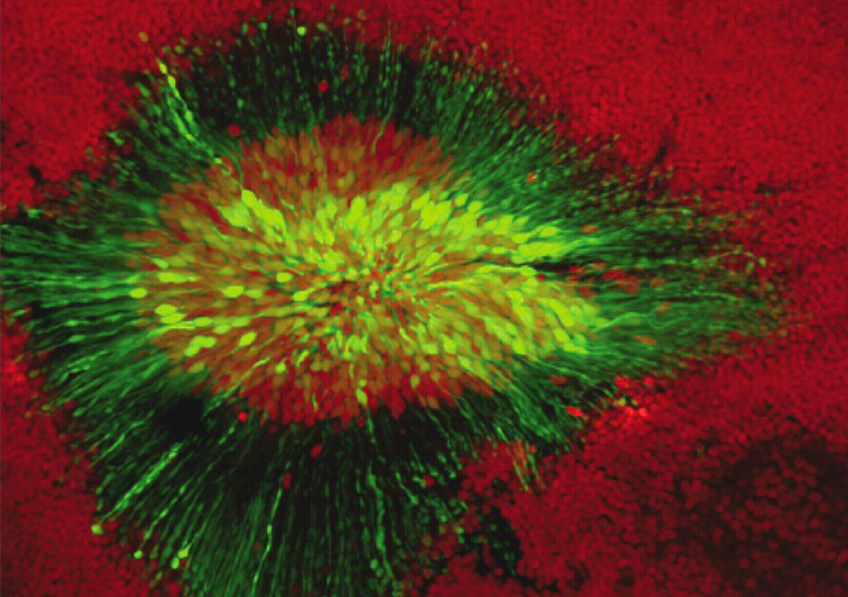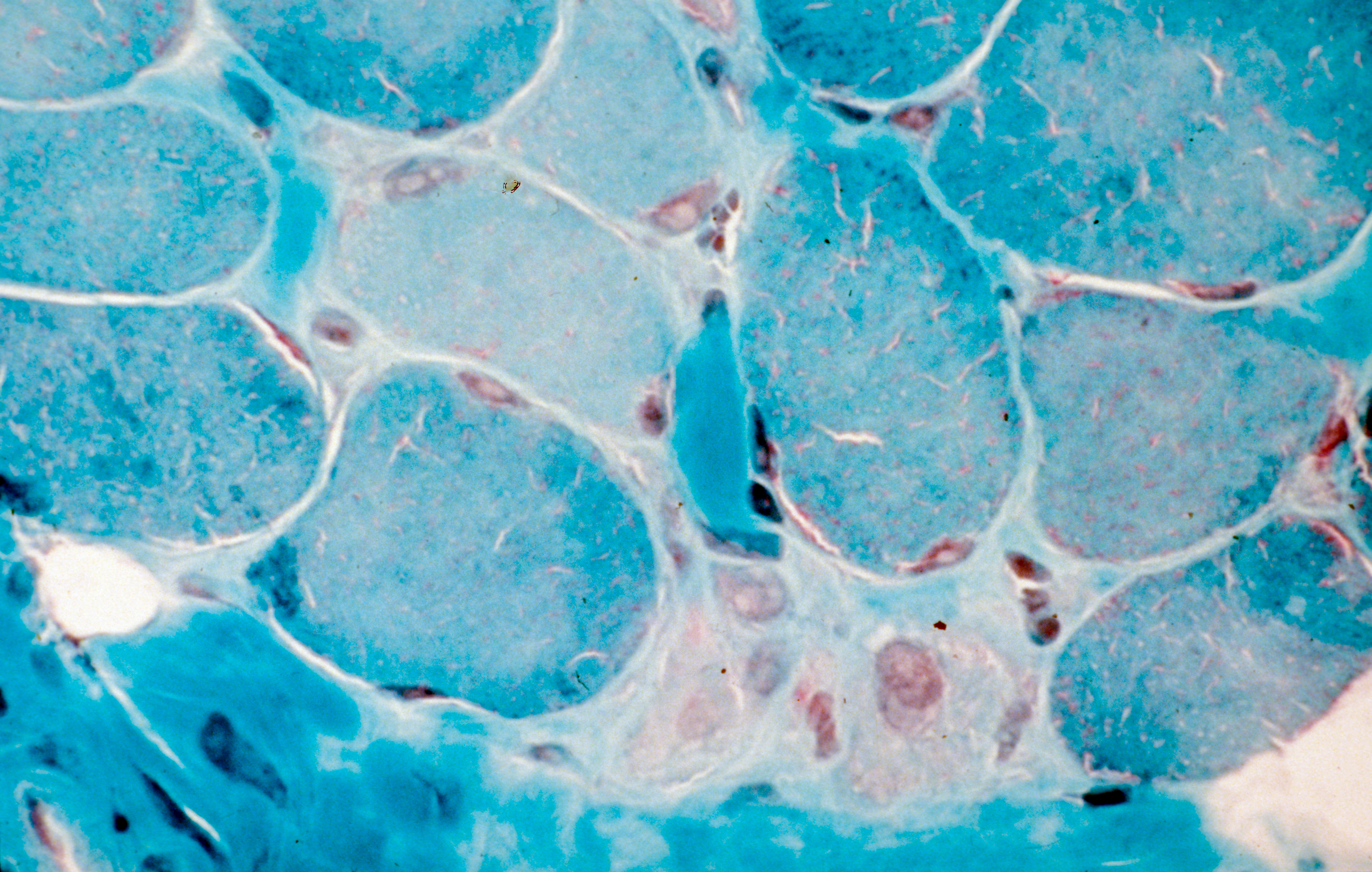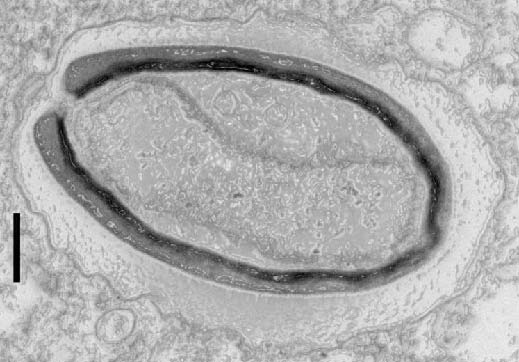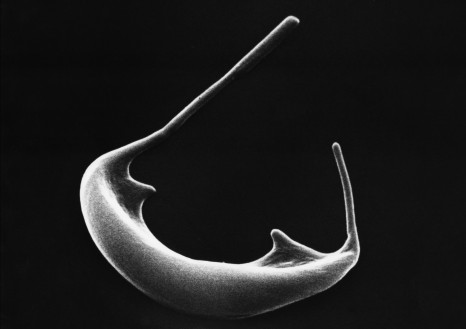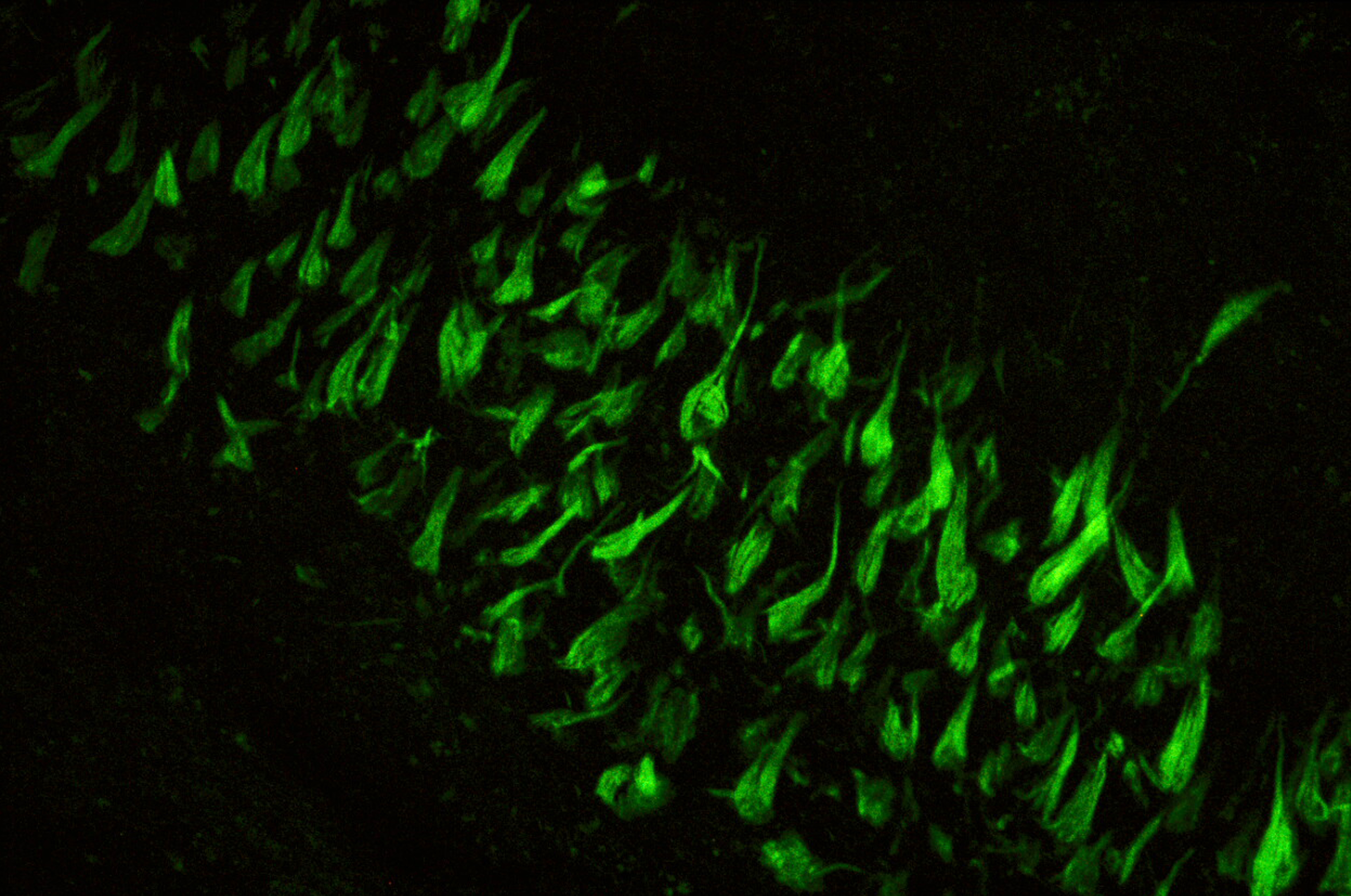
Agrégation de la protéine Tau dans la maladie d’Alzheimer. ©Inserm/U837, 2008
La propagation des agrégats de la protéine Tau dans le cerveau contribue à la progression de la maladie d’Alzheimer. Des chercheurs du Laboratoire des maladies neurodégénératives : mécanismes, thérapies, imagerie (CNRS/CEA/Université Paris-Sud, MIRCen), en collaboration avec l’Ecole normale supérieure, Sorbonne Université et l’Inserm, viennent d’identifier les cibles de ces agrégats. Publiés dans EMBO Journal le 10 janvier 2019, ces travaux permettront la conception d’outils capables de bloquer ces éléments clés dans la propagation des agrégats et de contrecarrer ainsi leur effet pathologique.
L’agrégation des protéines alpha-synucléine, pour la maladie de Parkinson, et Tau, pour la maladie d’Alzheimer, est intimement liée à la progression de ces pathologies neurodégénératives. Ces agrégats se propagent d’une cellule neuronale à l’autre en se liant aux cellules. Ils se multiplient[1] pendant cette propagation. Il a été montré que la propagation et l’amplification de ces agrégats protéiques sont délétères et contribuent à l’évolution de ces maladies.
La compréhension de la formation de ces agrégats, de leur propagation et de leur multiplication dans les cellules du système nerveux central présente un potentiel thérapeutique : elle permettrait de cibler ces processus et d’agir sur leurs conséquences.
Propagation des protéines
L’étape clé dans la propagation d’agrégats pathogéniques est la fixation d’agrégats provenant de cellules neuronales affectées aux membranes de cellules indemnes. Après avoir identifié les cibles des agrégats pathogéniques de la protéine alpha-synucléine (Shrivastava et al, 2015 EMBO J), l’équipe du Laboratoire des maladies neurodégénératives (CNRS/CEA/Université Paris-Sud, MIRCen, Fontenay-aux-Roses), en collaboration avec l’Ecole normale supérieure, Sorbonne Université et l’Inserm, vient d’identifier les cibles des agrégats de la protéine Tau. Il s’agit de la pompe sodium/potassium et des récepteurs du glutamate, deux protéines essentielles à la survie des neurones. L’expérience a été menée sur des neurones de souris en culture.
Modification des membranes neuronales
Les chercheurs ont également mis en évidence que les agrégats pathogéniques modifient la membrane des neurones en redistribuant les protéines membranaires. L’intégrité membranaire — et plus particulièrement celle des synapses, nœud de communication essentiel entre neurones — est affectée. Ces modifications sont délétères pour les neurones car elles entraînent une communication anormale entre eux ainsi que leur dégénérescence.
Ces travaux expliquent ainsi le dysfonctionnement précoce des synapses et la dégradation de communication normale observés dans les réseaux neuronaux au cours de l’évolution de la maladie.
Vers de nouvelles thérapies
Ils ouvrent aussi la voie à la conception de nouvelles stratégies thérapeutiques fondées sur la protection de l’intégrité synaptique, la restauration de l’activité des récepteurs membranaires de la protéine Tau et l’utilisation de leurres pour empêcher l’interaction délétère entre agrégats pathogènes de la protéine Tau et leurs cibles membranaires. Ces approches thérapeutiques pourront être menées à l’aide de neurones humains puisque les chercheurs du laboratoire viennent de développer ce type de cultures en collaboration avec le laboratoire I-Stem (Institut des cellules souches pour le traitement et l’étude des maladies oncogéniques, AFM-Téléthon/Inserm/Université Evry-Val d’Essonne) et Sorbonne Université. Cette dernière étude est également publiée le 10 janvier 2019, dans Stem Cell Reports[2].
[1] Ils s’amplifient en recrutant les protéines endogènes alpha-synucléine et Tau des cellules affectées pendant cette propagation
[2] Propagation of α-Synuclein strains within human reconstructed neuronal network. Simona Gribaudo, Philippe Tixador, Luc Bousset, Alexis Fenyi, Patricia Lino, Ronald Melki, Jean-Michel Peyrin, Anselme Louis Perrier, Stem Cell Reports, le 10 janvier 2019.
Le laboratoire rassemble près de 60 scientifiques dont les thèmes de recherche en neurosciences couvrent les mécanismes de dégénérescences, les modèles animaux, l’imagerie cérébrale, et l’étude de stratégies thérapeutiques géniques, cellulaires et médicamenteuses pour les maladies neurodégénératives, en particulier la maladie d’Alzheimer, la maladie de Parkinson et la maladie de Huntington.
Le LMN est situé au sein de MIRCen (Molecular Imaging Research Center) une installation de recherche préclinique développée par le CEA et l’Inserm. MIRCen est un des départements de l’institut de biologie François Jacob du CEA, sur le site de Fontenay-aux-Roses du CEA Paris-Saclay.