I
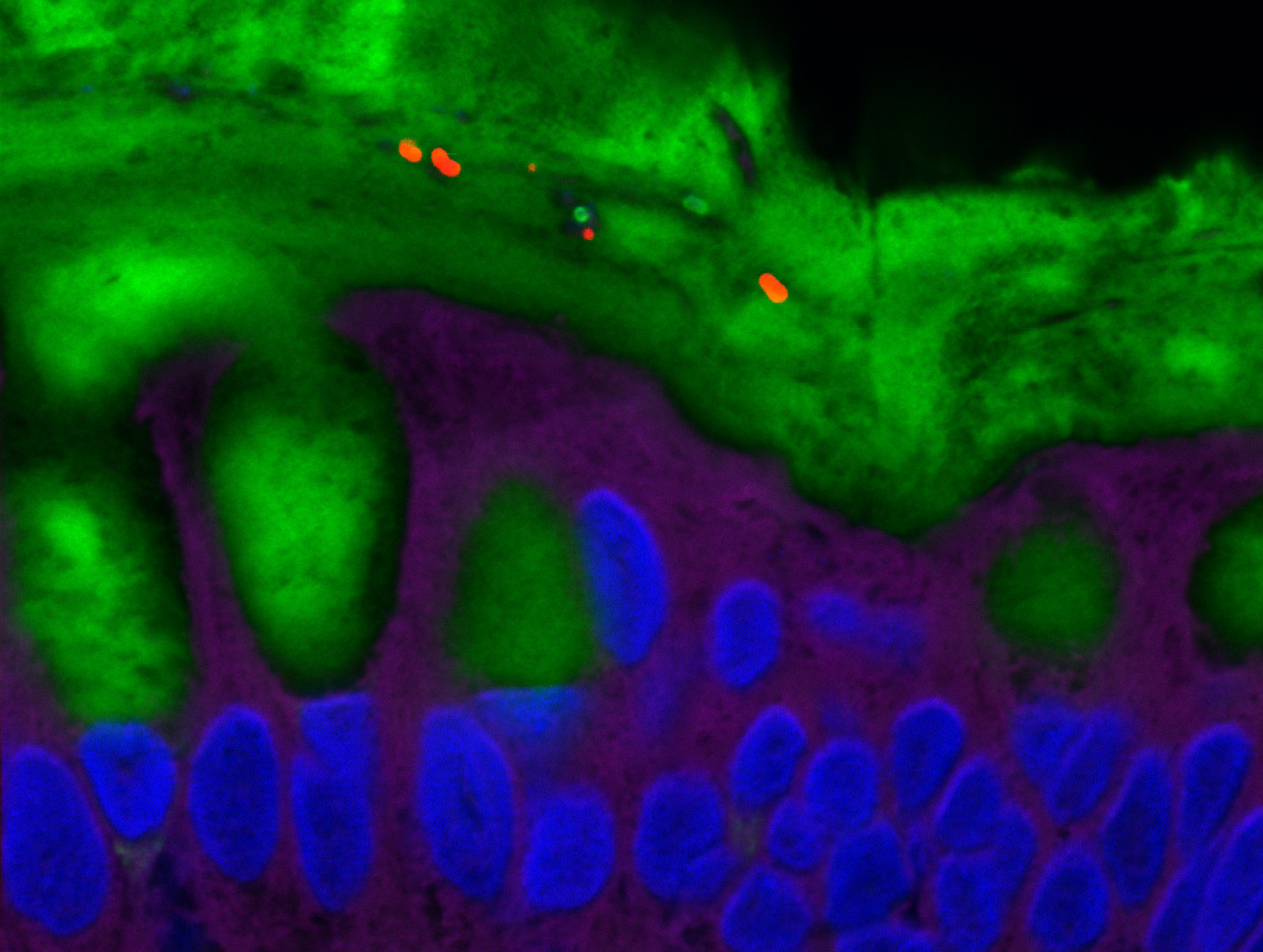
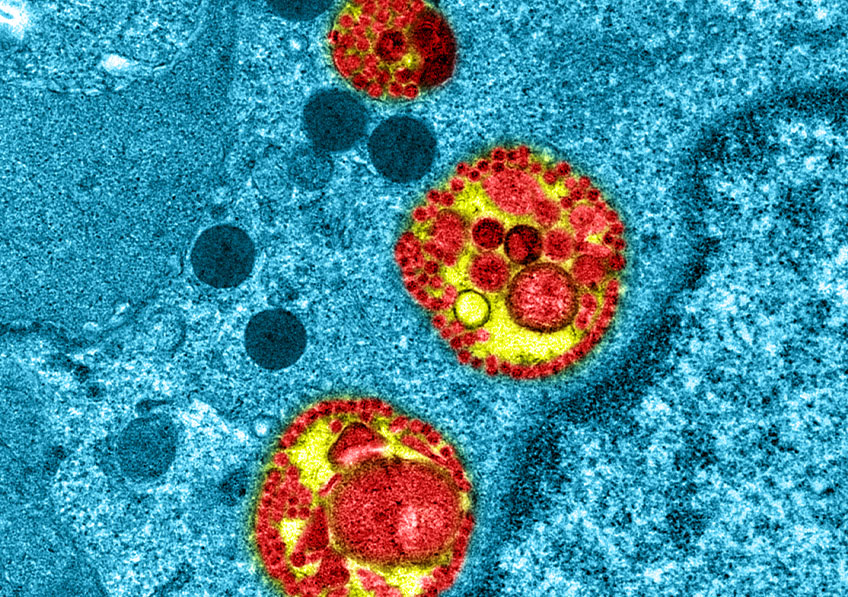
© Institut Pasteur. Image par Rémy Robinot, Mathieu Hubert, Vincent Michel, Olivier Schwartz & Lisa Chakrabarti, colors by Jean Marc Panaud
Des équipes de l’hôpital de la Pitié-Salpêtrière AP-HP, de Sorbonne Université, de l’Inserm et de l’Institut Pasteur ont mené des travaux pour étudier le rôle que jouent les anticorps de type IgA dans la protection de l’organisme contre la Covid-19 au niveau des muqueuses, notamment respiratoires. Ces travaux sous presse dans Science Translational Medicine, et qui font l’objet d’une pré-publication le lundi 7 Décembre 2020 sur le site de la revue Science Translational Medicine, montrent que la réponse des anticorps IgA joue un rôle primordial pour neutraliser le virus SARS-CoV-2 de manière précoce et particulièrement efficace.
Les anticorps de type IgA jouent un rôle essentiel dans la protection de l’organisme au niveau des muqueuses, notamment respiratoires. Il était donc logique d’étudier cette réponse anticorps particulière chez les patients infectés par le virus SARS-CoV-2. Les chercheurs et cliniciens affiliés au Centre de Recherche CIMI (Sorbonne Université et Inserm) en collaboration avec plusieurs services cliniques de l’APHP-Sorbonne Université et des équipes de l’Institut Pasteur, montrent que la réponse IgA joue un rôle primordial pour neutraliser le virus SARS-CoV-2 de manière précoce et particulièrement efficace.
D’une manière un peu surprenante, les anticorps IgA sont même souvent les premiers anticorps spécifiques du virus détectables. Un profil inhabituel puisque le dogme immunologique veut que la réponse IgM soit dominante lors de la rencontre avec un pathogène inconnu. La stimulation induite par le virus induit une expansion très importante de jeunes cellules sécrétrices d’anticorps IgA (plasmablastes) qui circulent entre le sang et les muqueuses au niveau desquelles elles sont capables de résider. Toutefois, cette réponse des anticorps IgA est lentement déclinante, y compris au niveau salivaire.
Les équipes en charge de ces travaux montrent que dans les premières semaines qui suivent l’infection, les anticorps de type IgA assurent la majeure partie du travail de neutralisation du virus.
Le taux de ces anticorps diminue rapidement dans le sang pour devenir faiblement détectable chez la plupart des sujets 30 jours après le début des symptômes. Ces anticorps restent toutefois plus longtemps détectables et actifs dans la salive (jusqu’à 73 jours après le début des symptômes) même si ce niveau de protection locale semble lui aussi décliner lentement. Chez un sous-groupe de patients ayant présenté une forme ambulatoire peu sévère, il est en effet rapporté qu’environ 6 mois après le comptage, la salive n’exerce plus de pouvoir neutralisant sur le virus. Ces résultats préliminaires sur la salive n’impliquent pas forcément une absence de protection au niveau tissulaire, surtout chez les patients ayant présenté une forme plus sévère de la maladie.En conclusion, ce travail met en évidence le caractère puissamment protecteur de l’IgA. Il pose la question du rôle possible de l’IgA sécrétoire dans la limitation de la transmission du virus.