Un consortium européen de chercheurs (1), dont les équipes françaises de Pierre Ronco, directeur de l’unité mixte de recherche 702 « Remodelage et réparation du tissu rénal » (UPMC / Inserm), Hanna Debiec (Inserm / UPMC) et de Bénédicte Stengel (Inserm / Univ Paris Sud UMR_S 1018) ont identifié des gènes de prédisposition à une maladie du rein : la glomérulonéphrite extramembraneuse. Difficile à traiter, celle-ci peut mener à une insuffisance rénale nécessitant le recours à la dialyse ou à la greffe. La découverte de ces gènes définit des biomarqueurs de la glomérulonéphrite extramembraneuse, ce qui pourrait considérablement améliorer la surveillance et la prévention de la maladie.
Les travaux des chercheurs, publiés dans la revue The New England Journal of Medicine datée du 17 février 2011, sont disponibles en ligne. Dans le même numéro, une lettre de Hanna Debiec et Pierre Ronco complète les aspects génétiques de la glomérulonéphrite extramembraneuse par des données immunologiques.
La glomérulonéphrite extramembraneuse est une maladie rare qui touche le rein. Dans 85 % des cas, ses causes sont indéterminées : elle est dite « idiopathique ». Dans cette maladie, certains anticorps, des immunoglobulines, se déposent dans les glomérules du rein, qui sont des structures sphériques formées d’anses capillaires servant à filtrer le sang et à produire l’urine. La paroi des capillaires et les cellules (podocytes) qui la tapissent, composent le filtre glomérulaire, qui va être « attaqué » par ces dépôts. Les lésions du filtre entrainent le passage anormal dans les urines des protéines de gros diamètre, comme l’albumine, et la diminution de la concentration de ces protéines dans le sang. Le sel et l’eau vont alors s’infiltrer dans les compartiments extracellulaires, provoquant des œdèmes. Dans les cas avancés, les glomérules présentent une fibrose importante, compromettant le fonctionnement du rein. A terme, la glomérulonéphrite extramembraneuse peut engendrer une insuffisance rénale grave qui, au stade terminal, nécessite le recours à la transplantation. Malheureusement, la maladie récidive dans près de 40 % des cas sur le rein greffé.
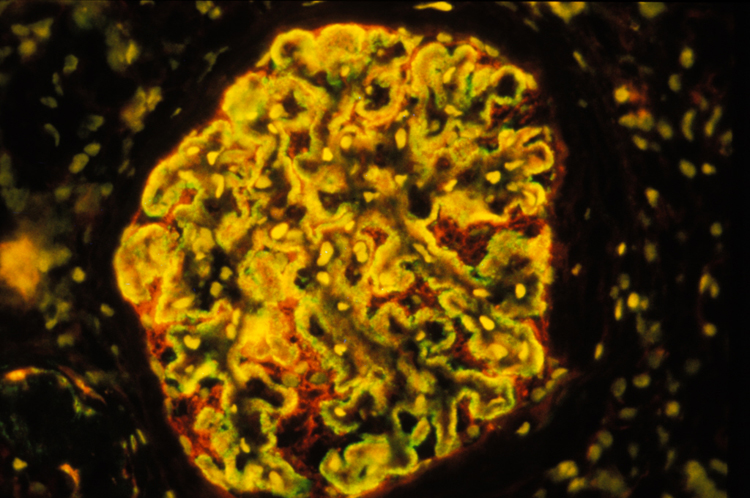
Néphron : coupe au cryostat dans un glomérule de rein humain chez un malade atteint de glomérulonéphrite extramembraneuse. Superposition des images 15050 et 15051. On met en évidence que les dépôts d’immunoglobulines sont à l’extérieur de la membrane basale, ce qui permet de caractériser cette maladie. © Inserm, Oriol, Rafael
Les mécanismes d’apparition de la glomérulonéphrite extramembraneuse sont encore mal connus. Hanna Debiec et Pierre Ronco ont cependant déjà dévoilé ceux d’une forme rare de glomérulonéphrite extramembraneuse néo-natale. En effet, ils ont étudié le cas d’un enfant né d’une mère atteinte d’un déficit en endopeptidase neutre, une enzyme normalement présente sur les podocytes du glomérule rénal. Les chercheurs ont découvert que la jeune femme a produit des anticorps contre cette enzyme, apportée pendant la grossesse par le placenta, et ils ont montré que les anticorps ont franchi la barrière placentaire, pénétré la circulation sanguine de l’enfant et atteint leur cible à la surface des podocytes, créant des dépôts extra-membraneux. Une autre équipe franco-américaine quant à elle, a récemment identifié chez 70% des adultes atteints de glomérulonéphrite, un autre antigène cible des anticorps sur les podocytes, le récepteur de la phospholipase A2 (PLA2R1). Contrairement à la glomérulonéphrite extramembraneuse de l’enfant d’origine allo-immune, la maladie de l’adulte est due à une réaction auto-immune.
Les équipes du Consortium ont exploré les bases génétiques de la maladie en étudiant de manière approfondie le génome de 556 malades adultes 398 hommes atteints d’une glomérulonéphrite extramembraneuse idiopathique et de près de 2 400 témoins. L’étude française a porté sur un groupe de sujets atteints de néphropathies glomérulaires, la cohorte GN-Progress, établie par Bénédicte Stengel et collaborateurs.
Les résultats montrent que le risque de développer une glomérulonéphrite extramembraneuse est 80 fois plus élevé chez les patients possédant 2 variants des gènes :
- PLA2R1, situé sur le chromosome 2q24,
- HLA-DQA1, situé sur le chromosome 6p21. Son rôle serait de présenter certaines parties (épitopes) de PLA2R1 au système immunitaire, qui en retour stimulerait la production d’anticorps contre PLA2R1.
Les résultats suggèrent en outre que des variations de séquence du gène PLA2R1 pourraient modifier les propriétés de cet antigène, influençant ainsi le développement de la réponse auto-immune.
Cependant, l’étude montre aussi que le risque de glomérulonéphrite extramembraneuse idiopathique est plus élevé avec les variants de HLA-DQA1 qu’avec ceux de PLA2R1, ce qui suggère que HLA-DQA1 pourrait favoriser le développement d’anticorps contre d’autres antigènes qui restent à identifier.
Ces résultats sont corroborés par la lettre publiée par Hanna Debiec et Pierre Ronco dans le même numéro de The New England Journal of Medicine, indiquant que 57% des sérums de patients atteints de glomérulonéphrite extramembraneuse « idiopathique » contiennent des anticorps anti-PLA2R1 alors que l’antigène PLA2R1 est retrouvé dans les dépôts immunologiques des glomérules chez les 3/4 des patients. La recherche d’autres cibles antigéniques de PLA2R1 s’avère donc nécessaire.
L’identification de ces 2 gènes de prédisposition à la glomérulonéphrite extramembraneuse idiopathique illustre parfaitement les découvertes récentes sur les mécanismes des maladies autoimmunes. En effet, celles-ci impliquent bien une « gâchette »-gène de réponse immune (ici HLA-DQA1), une « balle »-anticorps (ici des immunoglobulines anti-PLA2R1) et une « cible »-antigène, (ici l’antigène glomérulaire PLA2R1).
(1) Pierre Ronco et Hanna Debiec (Unité Inserm/UPMC UMR_S 702), Bénédicte Stengel (Unité Inserm/Univ Paris Sud UMR_S 1018), Centre National de Génotypage, Robert Kleta (University College de Londres, Angleterre), Peter Mathieson (Université de Bristol), Jack Wetzels (Radboud University, Nijmegen, Pays-Bas).