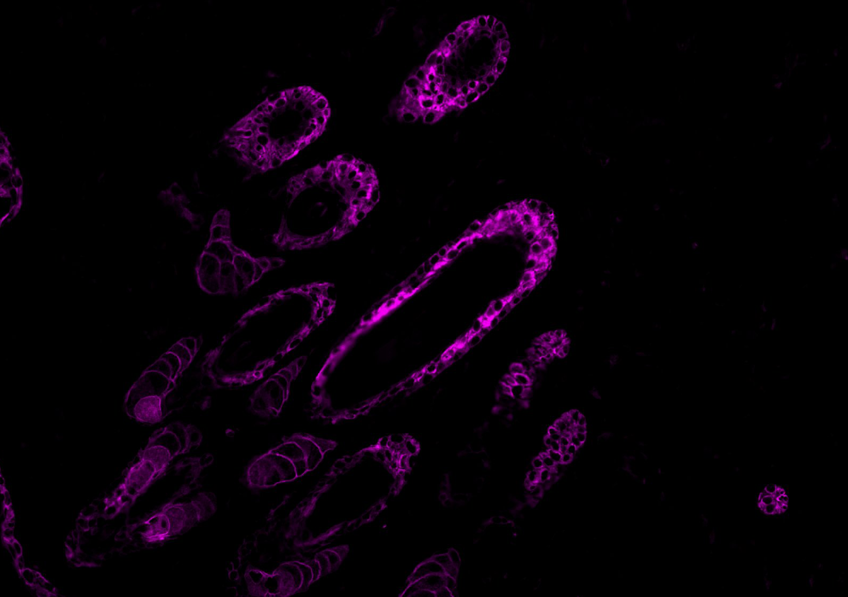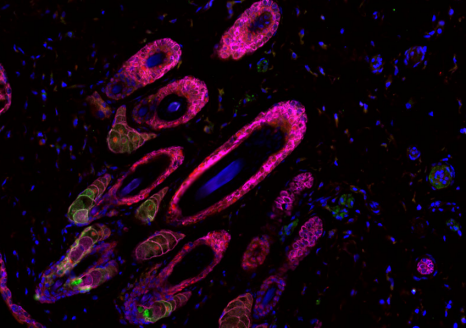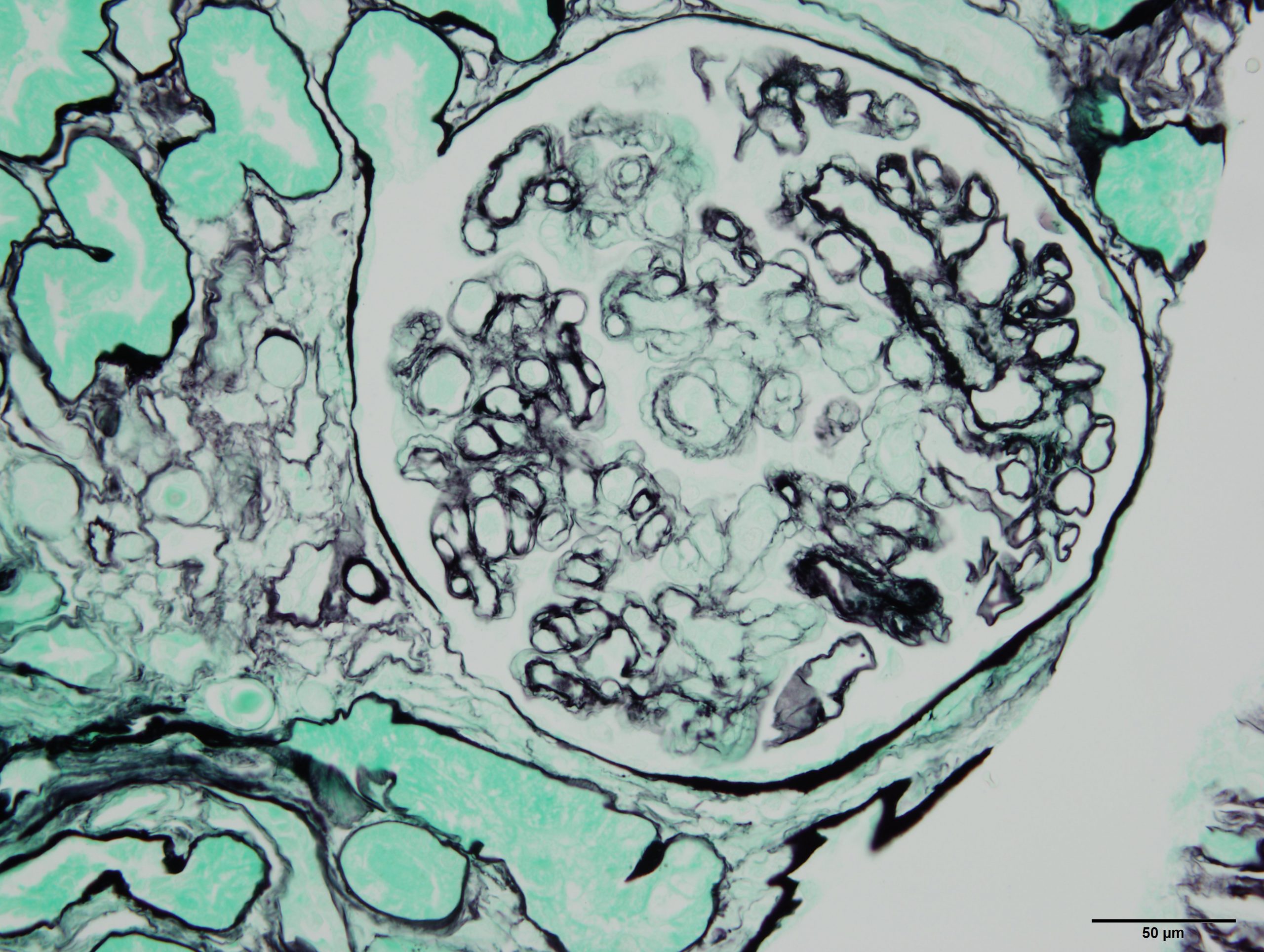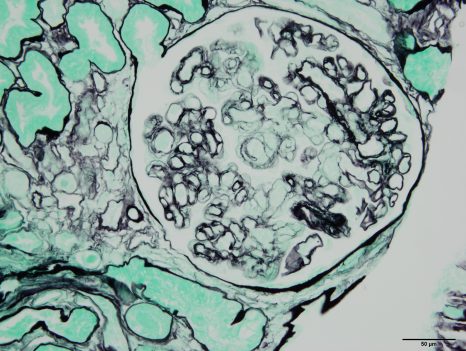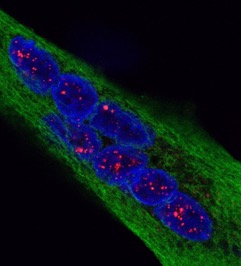
Transformation des cellules mammaires tumorales dans le cancer du sein. Crédits / Inserm – Xavier Coumoul
Des travaux décrivent le rôle épigénétique[1] d’un ARN non-codant dans le développement de tumeurs agressives, notamment dans le cancer du sein. L’étude, menée en collaboration entre l’Institut Curie, l’Inserm, le CNRS, l’Institut Paoli Calmettes, Aix-Marseille Université[2], vient d’être publiée dans la revue Cell. Ces résultats pourraient expliquer plus largement des biais de genre dans la prédisposition à certaines pathologies.
Tous les mammifères disposent de deux chromosomes sexuels. Les mammifères femelles possèdent deux chromosomes X, contrairement aux mâles qui ont un chromosome X et un Y. On connaissait déjà le rôle d’un ARN non-codant spécifique, appelé XIST, pour initier l’inactivation d’un des deux chromosomes X de la femelle. Le but de cette inactivation : bloquer la double expression des gènes situés sur ce chromosome car celle-ci affecte la viabilité des cellules. Dans cette nouvelle étude, les scientifiques démontrent que XIST joue non seulement un rôle pour déclencher cette inactivation du chromosome X mais aussi pour la maintenir tout au long de la vie des cellules.
Pour parvenir à ce résultat, les chercheurs et chercheuses ont étudié in vivo les effets de la suppression de XIST. Plusieurs techniques ont été utilisées pour cela. « Soit on a utilisé des outils génétiques pour bloquer l’expression de XIST, soit on a utilisé des techniques de CRISPR[3] pour interférer avec l’expression et on a rendu le gène de XIST silencieux », explique Raphaël Margueron, chercheur à l’Inserm et chef de l’équipe « Mécanisme de répression par les protéines Polycomb » à l’Institut Curie dans l’unité « Génétique et biologie du développement » (Institut Curie/CNRS/Inserm/Sorbonne Université).
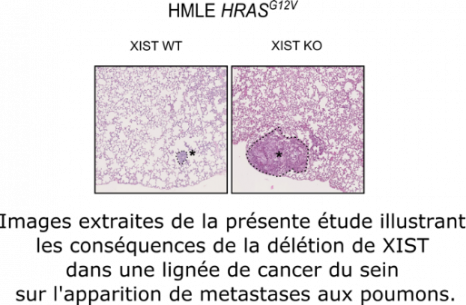
La perte de XIST dans les lignées cellulaires étudiées[4] a un effet important sur l’homéostasie[5] du tissu mammaire et impacte le développement tumoral. Raphaël Margueron précise que « quand on étudie des tumeurs et qu’on regarde après coup quelles étaient les propriétés de ces tumeurs, on voit qu’il y a une tendance à ce que XIST soit absent des tumeurs du sein les plus agressives. Ainsi qu’une réactivation d’un certain nombre de gènes du X inactif ».
Des gènes réactivés et la transcription s’emballe
Parmi les gènes réactivés par la perte de XIST, les chercheurs ont mis en évidence le gène codant pour MED14, une sous-unité essentielle au sein du complexe protéique Médiator. Celui-ci joue un rôle dans le contrôle de l’expression des gènes.
En conséquence, une augmentation de l’expression de MED14 va impacter l’activité de Médiator et contribuer à la perturbation de la différenciation des cellules souches mammaires[6]. Il s’agit potentiellement du résultat d’une augmentation de l’activation des enhancers (voir FOCUS ci-dessous).
En conclusion, la perte de XIST entraîne la réactivation de certains gènes (sur le chromosome X inactif) impliqués dans la différentiation des cellules et impacte le développement de cellules tumorales agressives. Ce mécanisme épigénétique étant spécifique à la présence de deux chromosomes X, ces résultats vont jouer un rôle majeur dans l’étude des prédispositions aux pathologies liées au genre de l’individu.
« Cette étude suggère que l’expression de XIST ainsi que de certains gènes liés au chromosome X pourraient être utilisés comme marqueurs de réponse à de nouvelles stratégies thérapeutiques », développe Christophe Ginestier, chef de l’équipe Inserm « Cellules Souches Epithéliales et Cancer » au Centre de recherche en cancérologie de Marseille.
Focus : Initiation de la transcription
« L’expression des gènes est contrôlée par les promoteurs mais aussi par des morceaux d’ADN, qui peuvent être assez distants du gène et du promoteur, qu’on appelle les enhancers. Il y a une communication entre les enhancers et les promoteurs. Le complexe Médiator intervient dans cette communication et permet aux enhancers de réguler finement l’expression des gènes. », explique Raphaël Margueron.
[1] L’épigénétique est une discipline qui étudie les mécanismes intervenant dans la régulation des gènes, essentielle à l’action des cellules et au maintien de leur identité.
[2] Les travaux ont été menés dans l’unité de recherche « Génétique et biologie du développement » (Institut Curie, CNRS, Inserm, Sorbonne Université) par l’équipe « Mécanisme de répression par les protéines Polycomb » de Raphaël Margueron ; au Centre de Recherche en Cancérologie de Marseille (CRCM / Inserm, CNRS, Aix-Marseille Université, Centre de Lutte Contre le Cancer de la région PACA-Institut Paoli-Calmettes) par l’équipe d’Emmanuelle Charaffe-Jauffret et de Christophe Ginestier et avec l’EMBL à Heidelberg (Edith Heard).
[3] La technique CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) consiste à interrompre ou suspendre l’expression d’un gène en le ciblant de manière précise.
[4] Le tissu mammaire contient des canaux composés de cellules basales et luminales. Les lignées cellulaires choisies permettent de reproduire cette hétérogénéité du tissu.
[5] Maintien de l’équilibre entre le milieu intérieur et extérieur.
[6] La différenciation est la capacité d’une cellule à acquérir une fonction propre. Une cellule souche peut devenir n’importe quelle cellule (musculaire, excrétrice, osseuse, etc.) mais c’est sa localisation (donc son environnement et les facteurs de transcription qu’on y trouve) qui va déterminer son devenir.